
Abstract
Autism spectrum disorder (ASD) is a complex neurodevelopmental condition with a strong genetic basis, and rare X-linked variants may contribute to its pathogenesis, particularly in consanguineous families. We present 2 pediatric cases from related Turkish families carrying a novel hemizygous DRP2 stop-gain mutation (p.Q232X). Case 1, a 5-year-old male, presented with ASD, macrocephaly, hypotonia, chronic otitis media with conductive hearing loss, and a mild demyelinating polyneuropathy confirmed electrophysiologically. Case 2, a 7-year-old male cousin, showed developmental delay, hyperactivity, bilateral cryptorchidism, and recurrent otitis media with hearing loss. Whole-exome sequencing identified the same DRP2 mutation in both patients, classified as likely pathogenic. Case 1 additionally carried heterozygous variants of uncertain significance in COL4A2 and MFN2. Multidisciplinary management included behavioral, speech, occupational, and physical therapies, as well as tympanostomy and orchiopexy. Longitudinal follow-up showed improved communication, motor function, and hearing, with stable neuropathy in Case 1. This report introduces a potentially novel role of DRP2 in ASD, extending its phenotypic spectrum beyond neuropathy, and underscores the need for further cases to define genotype–phenotype heterogeneity. Recognition of this variant is clinically relevant for genetic counseling in consanguineous populations, where risks extend beyond neuropathy to include broader neurodevelopmental outcomes.
Keywords
Introduction
Autism Spectrum Disorder (ASD) is characterized by deficits in social communication and interaction, along with restricted, repetitive patterns of behavior. 1 The etiology of ASD is multifactorial, involving a complex interplay of genetic and environmental factors. 2 Studies have implicated a wide array of genetic variants in the pathogenesis of ASD, ranging from chromosomal abnormalities to single-gene mutations.3 -5 However, the identification of novel genes associated with ASD remains critical for advancing our understanding of its molecular underpinnings and developing targeted interventions. Consanguinity, the union between individuals who are related as second cousins or closer, is prevalent in certain populations and increases the risk of autosomal recessive and X-linked recessive disorders.6,7 In consanguineous families, the probability of inheriting homozygous or hemizygous pathogenic variants is significantly elevated because of the sharing of common ancestors. This genetic background provides a unique opportunity to identify rare or novel genetic mutations that contribute to inherited diseases, particularly in isolated communities with limited genetic diversity.
The dystrophin-related protein 2 gene (DRP2) encodes a protein involved in the structure and function of myelinating Schwann cells in the peripheral nervous system. 8 Although mutations in DRP2 have been associated with neuropathies, their role in neurodevelopmental disorders such as ASD has not been well-established.9,10 To date, there are no published reports directly linking DRP2 variants to ASD phenotypes, making this observation potentially novel in the context of neurodevelopmental disorders. This study therefore aims to (i) describe the clinical and genetic findings of 2 pediatric cases with a hemizygous DRP2 stop-gain mutation, (ii) examine the phenotypic overlap of ASD with additional features such as macrocephaly, neuropathy, and cryptorchidism, and (iii) explore whether DRP2 dysfunction may represent an expanded pathway contributing to both peripheral nerve integrity and central neurodevelopment.
In this report, we present 2 pediatric cases from related consanguineous Turkish families, each exhibiting overlapping clinical features, including developmental delay, ASD, macrocephaly, and chronic otitis media. Both patients harbored a novel hemizygous stop-gain variant of DRP2 (p.Q232X). By reporting these findings, we seek to expand the genetic landscape of ASD, propose a potential extension of DRP2-related phenotypes beyond neuropathy, and emphasize the importance of genetic evaluation in patients with consanguineous backgrounds.
Case Description
Case History and Examination
The patient’s head circumference was 53 cm, which was above the 97th percentile for age and sex, indicating macrocephaly. He had a delayed closure of the anterior fontanelle, which remained partially open at 18 months. Neurological examination revealed mild hypotonia in the lower limbs with normal deep tendon reflexes. No signs of muscle atrophy or fasciculations were observed. Cranial nerve examination was unremarkable. No dysmorphic facial features or organomegaly was observed. Cranial MRI showed normal brain structures with no evidence of hydrocephalus, cortical malformations, or white matter abnormalities. The ventricles were normal in size and the corpus callosum was intact.
Electroencephalography (EEG) revealed normal background activity without epileptiform discharges or focal slowing. Electromyography (EMG) and nerve conduction studies were performed because of concerns regarding hypotonia and motor delays. The motor nerve conduction study, as detailed in Table 1, revealed increased distal latencies and decreased nerve conduction velocities in the median, ulnar, tibial, and peroneal nerves, indicating a mild demyelinating sensory-motor polyneuropathy. Sensory nerve conduction studies, presented in Table 2, showed reduced amplitudes and prolonged latencies in both the upper and lower extremities, supporting the presence of a sensory neuropathy component. The EMG findings summarized in Table 3 demonstrated reduced recruitment patterns and prolonged motor unit potentials, consistent with a neuropathic process affecting peripheral nerves.
Motor Nerve Conduction Study Results for Case 1.

Abbreviations: ms, milliseconds; mV, millivolts; mV·ms, millivolt milliseconds; mm, millimeters; m/s, meters per second; R, right.
Sensory Nerve Conduction Study Results for Case 1.

Abbreviations: ms, milliseconds; µV, microvolts; µV·ms, microvolt milliseconds; mm, millimeters; m/s, meters per second; P, phalanx; R, right.
Electromyography (EMG) Findings Summary for Case 1.
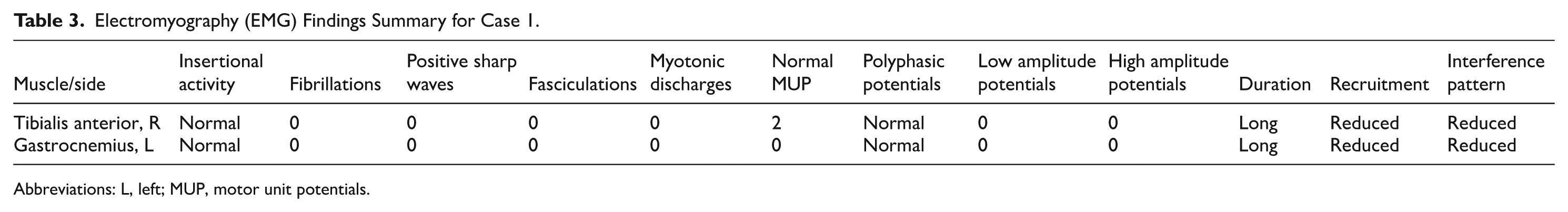
Abbreviations: L, left; MUP, motor unit potentials.
The patient had a history of recurrent otitis media since infancy and had experienced episodes every 2 to 3 months. Audiological evaluation included pure-tone audiometry, speech audiometry, and tympanometry. Pure-tone audiometry revealed bilateral conductive hearing loss with an average air-bone gap of 25 dB, more pronounced at low frequencies (500-1000 Hz). Speech audiometry showed a speech reception threshold (SRT) of 30 dB HL bilaterally with reduced speech discrimination scores (80% at 50 dB HL). Tympanometry indicated type B tympanograms consistent with middle ear effusion. He was diagnosed with chronic otitis media, which contributed to speech delays. Routine hematological and biochemical tests, including complete blood count, liver and renal function tests, and serum electrolytes, were within normal limits. Thyroid function test results were normal, excluding hypothyroidism as a cause of developmental delay. Metabolic screening, including plasma amino acids, urine organic acids, and acylcarnitine profile, did not reveal any abnormalities, reducing the likelihood of an inborn error of metabolism.
The patient had bilateral undescended testes for which he underwent surgical correction (orchiopexy) at ages 2 and 3. The postoperative recovery was uneventful. Neurological examination results were largely unremarkable, with normal muscle tone and reflexes. No signs of hypotonia or muscle weakness were observed. No dysmorphic features or organomegaly was observed. Cranial MRI showed normal findings with no structural abnormalities, cortical malformations, or signal changes. EEG demonstrated normal background activity without epileptiform discharges or focal abnormalities. The patient had a history of frequent ear infections and had experienced episodes every 2 to 4 months since infancy. He was diagnosed with chronic otitis media. Audiological assessment included pure-tone and speech audiometry. Pure-tone testing confirmed bilateral conductive hearing loss with an average air-bone gap of 30 dB, predominantly affecting low and mid frequencies (500-2000 Hz). Speech audiometry showed an SRT of 35 dB HL bilaterally with reduced speech discrimination scores of 75% at 55 dB HL. Tympanometry demonstrated type B curves, indicating persistent middle ear effusion. Hormonal evaluations, including luteinizing hormone (LH), follicle-stimulating hormone (FSH), and testosterone levels, were within normal limits, ruling out endocrine causes of cryptorchidism. Karyotype analysis was normal (46,XY), excluding chromosomal abnormalities. Routine laboratory tests, including complete blood count and metabolic panel, were unremarkable.
Differential Diagnosis
Based on clinical presentation, various genetic syndromes were considered. For both patients, Fragile X syndrome was evaluated due to developmental delays and behavioral issues. Rett syndrome and tuberous sclerosis complex were considered less likely due to the absence of characteristic features. Metabolic disorders, such as phenylketonuria and mucopolysaccharidoses, were also considered but deemed unlikely after normal metabolic screening results. For macrocephaly in Case 1, the differential diagnoses included Sotos syndrome and PTEN hamartoma tumor syndrome, but the absence of overgrowth features and normal MRI findings made these less probable. Cryptorchidism in Case 2 prompted evaluation for syndromes such as Kallmann syndrome and Prader-Willi syndrome; however, normal hormonal levels and the absence of other characteristic features reduced their likelihood. Given that testicular descent is a neurodevelopmentally regulated process dependent on genitofemoral nerve signaling, androgen action, and gubernacular response,11,12 variants affecting neuronal pathways such as DRP2 may plausibly contribute to aberrant descent mechanisms. This possibility, though speculative, suggests that cryptorchidism could represent an expanded phenotypic manifestation rather than a coincidental finding.
Genetic Investigations
Whole-exome sequencing (WES) was performed for both patients to identify potential genetic etiologies. Chromosomal microarray analysis did not reveal any pathogenic copy number variations, and Fragile X testing was negative, excluding the CGG repeat expansion in the FMR1 gene. In Case 1, WES identified a hemizygous stop-gain variant in DRP2(p.Q232X, c.694C > T), which was classified as likely pathogenic based on the American College of Medical Genetics and Genomics (ACMG) criteria (Figure 1). 13 This mutation introduces a premature stop codon, potentially resulting in a truncated protein or nonsense-mediated decay. Additionally, heterozygous variants of uncertain significance were detected in COL4A2 (p.Q1400X) and MFN2 (p.R707W), which may act as genetic modifiers. In Case 2, the same hemizygous stop-gain variant in DRP2 (p.Q232X) was identified, suggesting a shared genetic etiology within the family. All reporting followed the CARE (CAse REport) guidelines as recommended by EQUATOR to ensure methodological completeness. 14

Identification of the pathogenic DRP2 (p.Q232X) stop-gain mutation in 2 patients through whole-exome sequencing (WES).
Treatment and Management
Case 1: Early intervention was initiated with applied behavior analysis (ABA) therapy to address ASD symptoms, focusing on improving communication, social skills, and reducing repetitive behaviors. Speech and language therapy was provided to enhance language development and to improve expressive and receptive language skills. Occupational therapy targeted fine motor skills, coordination, and sensory integration to assist with daily activities. Physical therapy was implemented to address hypotonia and gait abnormalities, incorporating exercises to strengthen the muscles and improve balance. For audiological management, tympanostomy tubes were placed to manage chronic otitis media, which resulted in improved hearing thresholds and reduced infection frequency. Regular monitoring was scheduled with neurology, audiology, and developmental pediatrics to assess the patient’s progress and adjust the interventions as needed.
Case 2: Surgical management involved bilateral orchiopexy surgeries, which were successfully performed, with postoperative monitoring confirming a normal testicular position and size. Speech and language therapy was initiated to address expressive and receptive language delays using techniques to improve articulation and vocabulary. The patient was enrolled in an early childhood education program with individualized support to address learning difficulties and attention deficits. Audiological management included insertion of tympanostomy tubes to alleviate middle ear effusion, resulting in improved hearing and facilitation of speech development. Behavioral interventions were applied to manage hyperactivity and improve attention span, including behavior modification techniques and structured routines. Regular monitoring was continued with endocrinology to track testicular function and development as well as with audiology and developmental pediatrics to ensure overall progress.
Genetic counseling was provided to both families, emphasizing the X-linked inheritance pattern of the DRP2 mutation. The risks of recurrence in future offspring and the implications of consanguinity were discussed in detail. Female relatives were offered carrier testing to assess their risk status.
Outcome and Follow-Up
Case 1: Over 2 years of intensive therapy, the patient showed meaningful gains in social engagement and communication, progressing to simple sentence use and more reciprocal interactions. Motor function improved with occupational and physical therapy, permitting participation in age-appropriate activities such as running and climbing stairs. Following tympanostomy tube placement, otitis media frequency decreased and hearing thresholds stabilized. Neuropathy symptoms remained stable on serial neurological evaluations without evidence of progression.
Case 2: Speech and language therapy produced marked improvements in expressive language; the patient began constructing phrases, following multistep instructions, and engaging in basic conversations. Behavioral interventions reduced hyperactivity and improved attention, facilitating classroom participation. Audiological improvement followed tympanostomy with no further middle ear infections observed. Endocrine follow-up after orchiopexy confirmed normal testicular function. While cryptorchidism may be incidental, the possibility that it represents an expanded manifestation of DRP2 dysfunction merits further evaluation in additional cases.
Both patients continue multidisciplinary care with regular reassessments to tailor interventions. Families accepted genetic counseling and have been informed about X-linked inheritance and implications of consanguinity for future pregnancies. Given the small sample size, these outcome data are preliminary but indicate that early, coordinated intervention can yield functional gains even when a novel genetic variant is present.
Discussion
The identification of a hemizygous stop-gain variant in DRP2 (p.Q232X) in 2 related patients suggests a novel contribution of this gene to autism spectrum disorder (ASD) and related neurodevelopmental features. The variant introduces a premature termination codon and is therefore likely to cause nonsense-mediated mRNA decay or produce a truncated protein. Loss of DRP2 function may disrupt the periaxin–dystroglycan complex in Schwann cells, impairing myelination and plausibly explaining the sensory–motor polyneuropathy observed in Case 1. 10 Crucially, the presence of core ASD features in both patients supports a conceptual extension of DRP2 pathology from peripheral nerve integrity to central neurodevelopmental regulation. This conceptual extension of DRP2 pathology from peripheral neuropathy to central neurodevelopmental dysfunction represents the innovative aspect of our report.
Macrocephaly in Case 1 broadens the phenotypic spectrum associated with DRP2 variants; macrocephaly is described in a subset of ASD cases and may reflect altered neuronal growth or connectivity. In Case 2, cryptorchidism could represent an additional phenotypic feature because testicular descent depends on both hormonal signaling and intact genitofemoral nerve pathways.15 -17 Nevertheless, coincidence remains possible; the clinical differences between the 2 cases suggest phenotypic heterogeneity and therefore indicate that more cases are required to define the spectrum of DRP2-associated presentations.
Case 1 also harbored heterozygous variants of uncertain significance in COL4A2 and MFN2, which may act as genetic modifiers. COL4A2 has been linked to cerebral small-vessel disease and porencephaly, 18 and MFN2 to Charcot–Marie–Tooth disease type 2A, 19 so coexisting variants could plausibly influence clinical severity or expressivity, although this remains speculative without functional data.
Consanguinity likely facilitated identification of this X-linked hemizygous variant. The male-limited phenotype aligns with X-linked recessive inheritance and potential lyonization in carrier females. Genetic counseling in such families should extend beyond discussion of neuropathy risk to include the possibility of broader neurodevelopmental outcomes, a consideration of particular importance in populations with high rates of consanguinity.
Whole-exome sequencing (WES) was decisive after standard testing (Fragile X and metabolic screens) was uninformative. WES can identify rare, clinically actionable variants and should be considered early in complex neurodevelopmental presentations, particularly in consanguineous pedigrees. Early multidisciplinary interventions in both cases were associated with functional improvements, reinforcing the value of timely diagnosis and integrated care.20 -22
The small number of related cases limits causal inference, yet the phenotypic variability observed between the 2 patients suggests that DRP2-associated disease may represent a broader and more heterogeneous spectrum than previously appreciated. This observation provides a basis for future work, where larger case series, international registries, and functional studies will be required to clarify genotype–phenotype correlations. Experimental approaches, including cellular and animal models of the p.Q232X variant and systematic screening of neurodevelopmental cohorts, may help determine whether DRP2 defines a reproducible syndromic entity.
Conclusion
This case report introduces a novel association between a hemizygous DRP2 stop-gain mutation and ASD, expanding the gene’s phenotypic spectrum to include macrocephaly and possibly genitourinary involvement. While causality cannot be inferred from 2 familial cases, the findings provide a signal warranting further study. Recognition of this variant has immediate implications for genetic counseling in consanguineous populations, where counseling should address risks that extend beyond neuropathy to include broader neurodevelopmental outcomes. Larger series, functional validation, and registry-based approaches will be needed to confirm whether DRP2 defines a reproducible syndromic entity.
Footnotes
Acknowledgements
The authors extend their gratitude to Medicos In Research, Nautanwa, Uttar Pradesh, 273164, India, an initiative by Dr. Amogh Verma, for their unwavering guidance and support throughout the development of this manuscript. Their invaluable contributions provided a platform for us to learn research methodologies, collaborate effectively, expand our professional network, and gain additional insights that significantly enriched this work. We deeply appreciate their efforts in fostering an environment of academic growth and scientific inquiry. The authors would also like to thank the patients and their family members for their cooperation in contributing to the advancement of the scientific community and extend their gratitude to the Intergen Genetic Center, Turkey, for conducting the genetic analysis.
Ethical Considerations
Ethical approval was not required for this study.
Consent to Participate
Written informed consent was obtained from the legal guardians of both patients in accordance with the journal’s policies.
Author Contributions
● Hajira Karim: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Resources, Data Curation, Writing – Original Draft, Visualization.
● Lima Oria: Conceptualization, Visualization, Writing – original draft, Writing – review & editing.
● Aslı Güner Öztürk Demir: Conceptualization, Visualization, Writing – original draft, Writing – review & editing.
● Muhsin Elmas: Conceptualization, Visualization, Writing – original draft, Writing – review & editing.
● Amogh Verma: Methodology, Software, Validation, Formal analysis, Resources, Data Curation, Supervision, Project administration, Writing – Original Draft, Writing – Review & Editing,
Funding
The authors received no financial support for the research, authorship, and/or publication of this study.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this study.
Guarantor
Amogh Verma.
Declaration of Generative AI and AI-Assisted Technologies in the Writing Process
Paperpal and ChatGPT-4o, were utilized solely for language, grammar, and stylistic refinement. These tools had no role in the conceptualization, data analysis, interpretation of results, or substantive content development of this manuscript. All intellectual contributions, data analysis, and scientific interpretations remain the sole work of the authors. The final content was critically reviewed and edited to ensure accuracy and originality. The authors take full responsibility for the accuracy, originality, and integrity of the work presented.
Data Availability Statement
Data sharing is not applicable to this article, as no datasets were generated or analyzed during the current study. All the information is available within the manuscript (including images). Any additional information can be obtained by contacting the corresponding author upon reasonable request.