
Abstract
Background:
Primary deficiency of coenzyme Q10 deficiency-4 (CoQ10D4) is a heterogeneous disorder affecting different age groups. The main clinical manifestation consists of cerebellar ataxia, exercise intolerance, and dystonia.
Case report:
We provide a case of adolescence-onset ataxia, head tremor, and proximal muscle weakness accompanied by psychiatric features and abnormal serum urea (49.4 mg/dL), lactate (7.5 mmol/L), and CoQ10 level (0.4 µg/mL). Brain-MRI demonstrated cerebellar atrophy, thinning of the corpus callosum, and loss of white matter. Whole exome sequencing showed a homozygous missense mutation (c.911C>T; p.A304V) in CoQ8A gene which is a rare mutation and responsible variant of CoQ10D4. After supplementary treatment with CoQ10 50 mg/twice a day for 2 months the clinical symptoms improved.
Conclusion:
These observations highlight the significance of the early diagnosis of potentially treatable CoQ8A mutation as well as patient education and follow-up. Our findings widen the spectrum of CoQ8A phenotypic features so that clinicians be familiar with the disease not only in severe childhood-onset ataxia but also in adolescence with accompanying psychiatric problems.
Introduction
Primary coenzyme Q 10 (CoQ10) is a group of inherited mitochondrial disorders characterized by multisystem and diverse clinical features including cerebellar ataxia, exercise intolerance, muscle weakness, cardiomyopathy, and nephropathy.1,2 The onset of symptoms and severity of manifestations may be from infancy till adulthood with severe to mild presentation, respectively.1-3 Several genes coding for proteins in CoQ synthesis are involved in the pathogenesis of this disease which interferes with CoQ10 function in the oxidative phosphorylation system. To date, mutations in ten genes (CoQ2, CoQ4, CoQ5, CoQ6, CoQ7, CoQ8A, CoQ8B, CoQ9, PDSS1, and PDSS2) were reported. 4 Primary deficiency of coenzyme Q10 deficiency-4 (CoQ10D4) is an autosomal recessive disorder caused by mutations in the CoQ8A gene. Similar to the yeast ABC1 protein, the human CoQ8A gene (also known as the AarF domain containing kinase 3 gene [ADCK3]) encodes a mitochondrial protein that has kinase-like and ATPase activity in the respiratory chain. 4 Patients may experience pure ataxia 5 or a progressive course of ataxia in addition to seizure, intellectual disability, exercise intolerance, and psychiatric symptoms.4,6,7 Less common manifestations include writing or speech coordination difficulties. 7 Early treatment with supplemental CoQ10 often halts the disease progression and improves patients’ symptoms which signifies prompt diagnosis. 2
We encountered a patient with adolescence-onset primary CoQ10 deficiency owing to a rare variant of the CoQ8A gene mutation (c.911C>T; (p.A304V)) who presented with cerebellar ataxia, exercise intolerance, and proximal muscle weakness. We discuss the clinical outcomes and course, with a focus on the necessity of patient education, early diagnosis, and the necessity for follow-up. Besides in order to identify the clues that predict potential treatment response we reviewed different variants of CoQ8A reported in the literature.
Case Presentation
A 12-year-old female child was referred to the pediatric clinic due to progressive ataxia, tremor of head, and anorexia. Although the patients’ symptoms have started at the age of 10 years with unsteady gait and tremor, no admission is obtained till the severity of symptoms. She was the only child from a healthy non-consanguineous parent who was born at 37 weeks through normal vaginal delivery (NVD) that had normal birth (Birth weight = 3700 g, Height = 50 cm, Head circumference = 33 cm) and development.
The past medical history was remarkable for mitral valve prolapse (MVP) (diagnosed confirmed via echocardiography) and irritable bowel syndrome (IBS) for which propranolol 10 mg was taken. Recurrent and more frequent infections of ear and throat during these 2 years are also reported for which the patient received amoxicillin 500 mg every 8 hours or azithromycin 2 g single dose. The history of seizure episodes was negative. No notable family history of similar symptoms was found.
The vital signs were as follows; pulse rate: 105 rate/minute, blood pressure: 110/70 mmHg, temperature: 36.9°C, respiratory rate: 14 rate/minute, O2 Saturation: 98%. The examination showed no facial or skeletal dysmorphia. Mild to moderate dysarthria, head tremor, bilateral dysmetria, and ataxic gait were positive neurological findings in the physical exam. Besides, finger-to-nose, and heel-to-shin tests were abnormal. Eye examination did not show Kayser-Fleischer rings. The visual and auditory acuity were normal. The motor and sensory neuron systems showed normal development and deep tendon reflexes were normal. Babinski’s sign was absent. No telangiectasia was observed. Psychiatric examination represents a spectrum of anxiety and depression (mild to moderate according to DSM-5). The Wechsler intelligence test was normal.
Laboratory analyses are depicted in Table 1. The results showed a high lactate level (7.5 mmol/L, reference range [RR]: 0.5-1.6 mmol/L) and low CoQ level (0.4 µg/mL, RR: 1.31 ± 0.38 µg/mL). Blood urea level was elevated (49.4 mg/dL, RR: 15-36 mg/dL). Thyroid function tests were normal. Vitamin A, E, and B complex, Ceruloplasmin, copper, and autoantibodies levels were checked and all were in the normal range. 25 (OH) Vitamin D was deficient (<8 ng/mL, RR is mentioned in Table 1). Urine analysis was positive for ketone. Urine culture and stool exam were normal. Cerebrospinal fluid (CSF) analysis was not obtained since the patient and her parents did not consent to lumbar puncture. Electromyography (EMG) and nerve conduction study (NCS) were normal. However, muscle biopsy showed lipid droplets.
Laboratory findings.
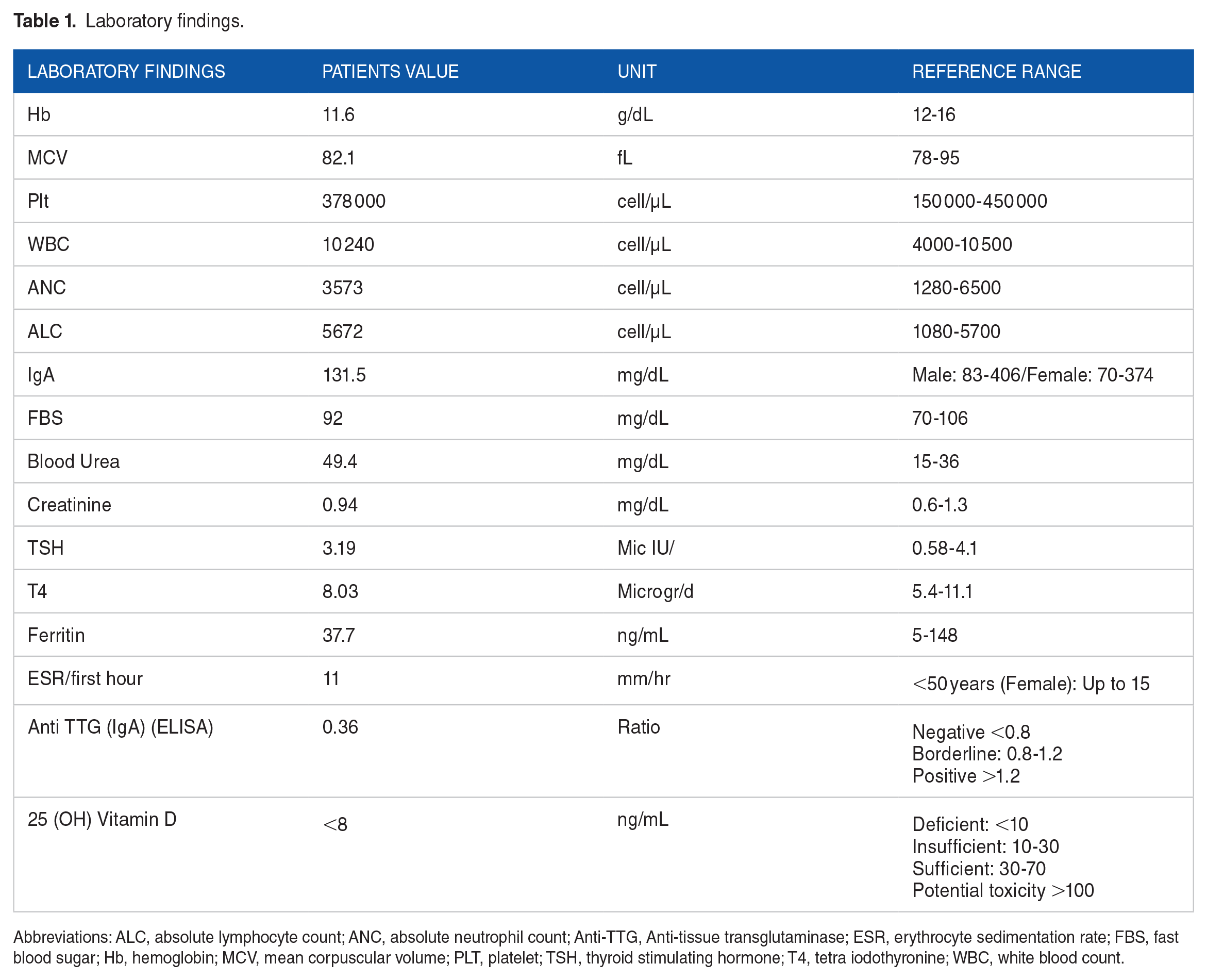
Abbreviations: ALC, absolute lymphocyte count; ANC, absolute neutrophil count; Anti-TTG, Anti-tissue transglutaminase; ESR, erythrocyte sedimentation rate; FBS, fast blood sugar; Hb, hemoglobin; MCV, mean corpuscular volume; PLT, platelet; TSH, thyroid stimulating hormone; T4, tetra iodothyronine; WBC, white blood count.
The patient was referred to pediatric neurologist at this time for further evaluation. Electroencephalogram (EEG) was performed and was normal. However, cranial magnetic resonance imaging (MRI) suggested pan-cerebellar atrophy, thinning of the corpus callosum, and white matter loss as depicted in Figure 1.

Magnetic resonance imaging showing pan-cerebellar atrophy (white arrow), thinning of corpus callosum (yellow arrow), and loss of white matter (red arrow).
Whole exome sequencing (WES) was done on DNA samples from the patient and her parents’ fibroblasts. Confirmatory tests were done using Sanger sequencing methods. A homozygous missense mutation for cytosine to thymidine at nucleotide 911 of CoQ8A (ENST00000366777.4: c.911C>T; [p.A304V]) was detected.
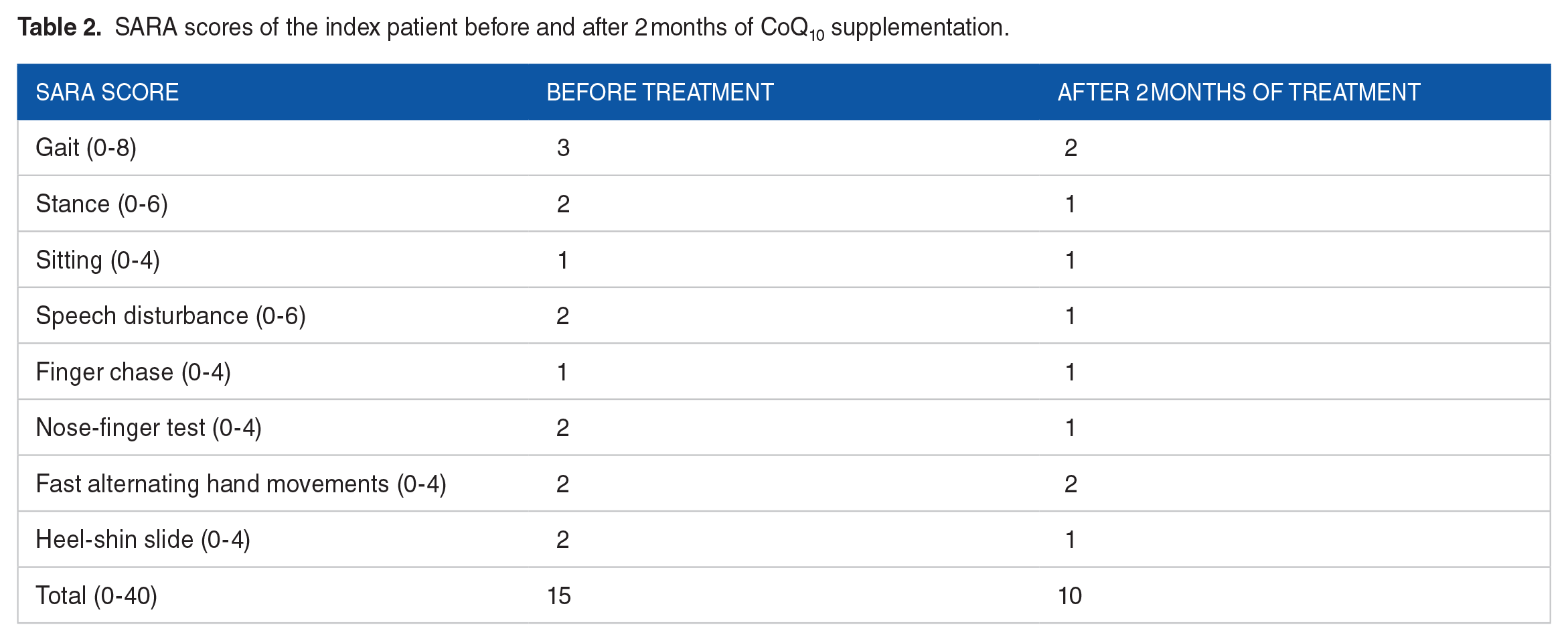
A mitochondrial disorder is suspected (MIM: 612016) and treatment with CoQ10 50 mg twice daily started on. The patient underwent follow-ups every 2 months while receiving CoQ10. After 2 months the symptoms improved and the progression is halted as shown by Scale for the Assessment and Rating of Ataxia (SARA) score (15 at baseline; and 10 after 2 months of treatment) (Table 2). The most noticeable improvement was the resolution of tremor. The spinocerebellar degeneration functional scale (SDFS) was improved after 2 months of treatment (5/7 at baseline; and 2/7 after 2 months of treatment). Infections of ear and throat have no longer recurred. After treatment, the serum abnormality in lactate and CoQ status was resolved. The serum lactate decreased to 1.2 mmol/L (reference range [RR]: 0.5-1.6 mmol/L) and CoQ level increased to 1.8 µg/mL (RR: 1.31 ± 0.38 µg/mL).
SARA scores of the index patient before and after 2 months of CoQ10 supplementation.
In the last follow-up, the patient was advised to refer to psychiatrist for her concern about lack of self-esteem in the school due to unstable walking and less memorizing ability compared with other students. She also reported fear of being in crowded places.
Discussion
Mutations in CoQ8A gene cause CoQ10D4, also known as spinocerebellar ataxia-9 (SCAR9) or autosomal recessive cerebellar ataxia type 2 (ARCA2).8,9 Patients with pathogenic variants of CoQ8A present with variable symptoms including gait ataxia, dystonia, seizure, exercise intolerance, and cognitive disabilities. The age of onset and severity of clinical manifestations varies.8,10 To date, 64 patients with 46 CoQ8A mutations have been reported. Most reported manifestations started from infancy and early childhood. Less but not least symptoms initiated in older ages. To be more precise, in this study we decided to categorize the age at which first symptoms occurred into 4 groups:
(1) Infancy (0-1 year old)
(2) Childhood (>1-9 years old)
(3) Adolescence (10-19 years old)
(4) Adult (⩾20 years old)
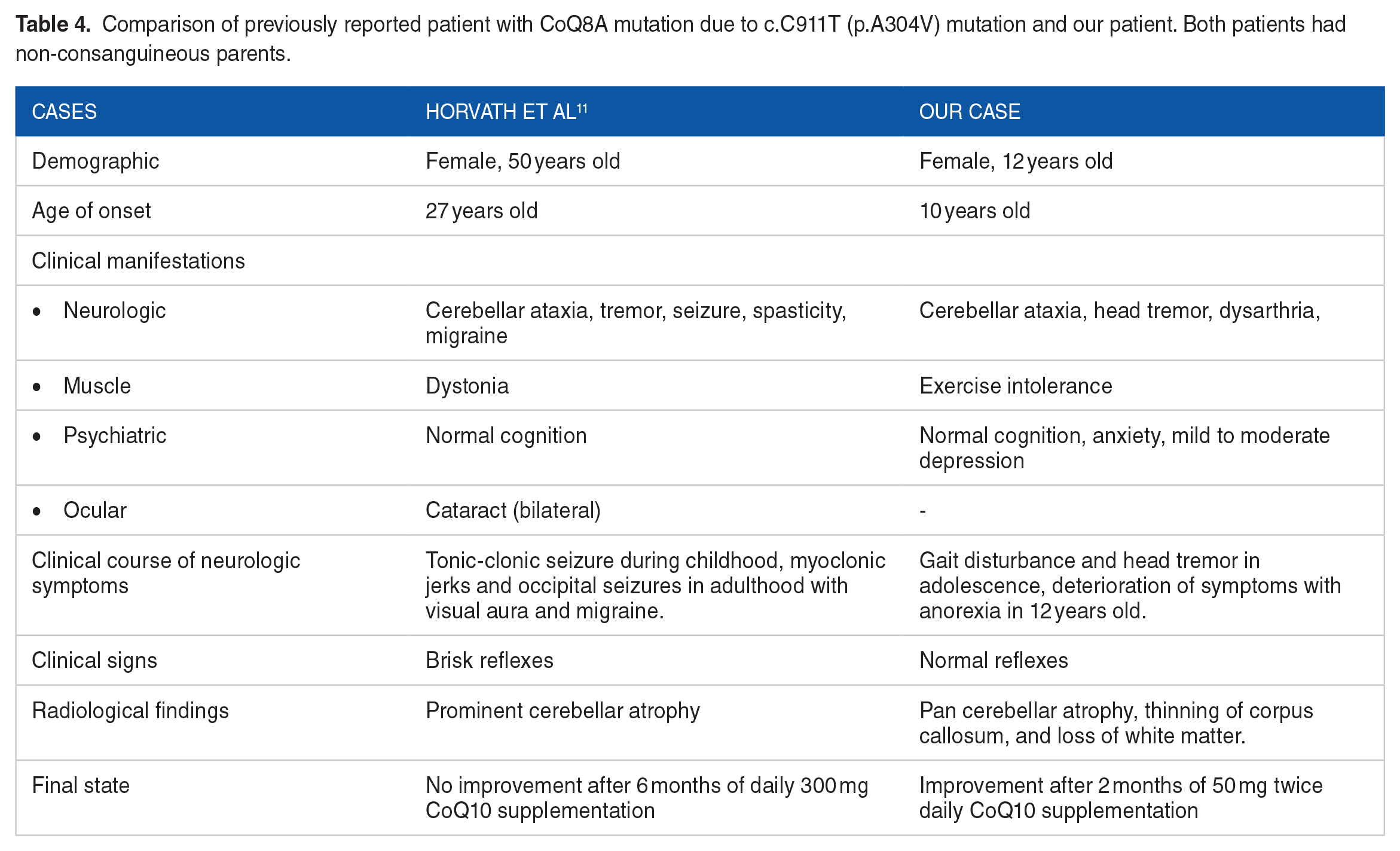
The reported pathogenic and likely pathogenic variants are summarized in Table 3. Our patient had homozygous missense mutation c.C911T; (p.A304V). Only one case with a similar genetic result was previously reported by Horvath et al. 11 The similarities and differences between the reported patient and our case are demonstrated in Table 4. Our patient is unique due to earlier onset (adolescence onset) of symptoms (10 years of age with ataxic gait and head tremor) as well as improvement of symptoms with treatment. Other notable laboratory features of our case were high ketonuria and high serum urea level. According to the literature, renal impairment ranging from proteinuria to end-stage renal disease (ESRD) may be seen in patients with CoQ10 deficiency. However, high urea level was reported in CoQ212-15,CoQ6,16,17 and PDSS2 deficiency. 18 Based on our knowledge, none of the previously reported patients with mutations in CoQ8A gene suffered from renal disease.
Pathogenic and likely pathogenic variants, age range, and response to treatment of previously reported patients with CoQ8A gene mutation.

days, months and years are abbreviated as d, m, y, respectively.
Abbreviations: BD, 2 times a day; TDS, 3 times a day.
Comparison of previously reported patient with CoQ8A mutation due to c.C911T (p.A304V) mutation and our patient. Both patients had non-consanguineous parents.
CoQ10 plays a crucial role in the mitochondrial respiratory chain which is responsible for generating ATP. The pathogenesis of CoQ10 deficiency is related to energy deficiency and lack of antioxidant defenses. 25 It is not yet clear through what mechanism the detected missense mutation (c.911C>T; [p.A304V]) caused an impairment in the function of CoQ10 biosynthesis. This pathological variant, which is functionally validated by segregation and CoQ level, 30 is thought to affect the amino acids in the UbiB protein kinase-like family. 31
Psychiatric problems in patients with mitochondrial disorders were previously reported in the literature.32,33 Depression is one of the frequently reported associated symptoms of patients with CoQ8A mutation.7,11,25 A multicenter study conducted by Traschütz et al reported psychiatric features such as anxiety, psychotic symptoms, depression, and aggression in about 25% of patients with CoQ8A mutation. 34 Mancuso et al investigated conditions such as agoraphobia, panic disorder, major depressive disorder, and social anxiety disorder in about 20% to 25% of patients with mitochondrial disease. 33 Also, some studies revealed the amelioration of psychiatric symptoms with CoQ10 supplementation35,36 and deterioration of underlying psychiatric problem with cessation of the mentioned treatment. 22 However, the exact pathophysiology and therapeutic efficacy of using CoQ10 remained unclear. In current study the patient suffered from low self-esteem, memory problems and fear of crowds. These symptoms did not improve after 2 months of supplemental treatment. Hence, this study aims to encourage psychiatric evaluation and follow-up in individuals with adolescence onset CoQ10 deficiency both before and after treatment.
The most common radiological finding in patients with CoQ8A is cerebellar atrophy that can be in different patterns (either localized, 4 diffused 27 , or pan-cerebellar 23 ). Other neuro-imaging radiological findings consist of cerebral and brainstem atrophy, stroke-like signal changes, infra-tentorial T2 hyperintensities, thin corpus callosum, enlarged ventricles, basal ganglia involvement, and thoraco-lumbar scoliosis.11,19,21,34 Herein, we found pan-cerebellar atrophy, thinning of corpus callosum, and loss of white matter as well as cerebellar atrophy. Although the MRI is the most important diagnostic method, Diffusion tensor imaging (DTI) and fiber tractography (FT) to reveal changes of fiber tracts 37 or phosphorus magnetic resonance spectroscopy imaging (P-MRSI) to monitor high-energy metabolites have been used in some cases. 37 The latter can also be used as a marker for mapping the treatment response. However, technical hurdles such as low sensitivity, long acquisition time and low signal have limited the utilization in clinical settings. 38
It is previously investigated that the treatment response is not associated with the age of onset, age and disease duration at the time of treatment initiation, SARA score, cumulative daily dose of CoQ supplementation, and the type of mutation. 34 We observed improvement in disease symptoms measured by SARA scores, SDFS, and improved lactate and CoQ10 serum levels. This is while in the study or Horvath et al with a similar mutation to our case, no improvement was reported after 6 months of treatment even with higher doses. 11 We hypothesized that the difference in treatment effectiveness may be due to different neurological disability stages.28,31 Spinocerebellar degeneration functional score (SDFS) is a rating scale to evaluate the disability stage that spans from 0 (no disability) to 7 (bedridden). The SDFS was significantly different between treatment responders in the study of Traschütz et al which was consistent with current study. 34 However, still larger studies are required to identify an accurate predictive factors.
There was a significant delay in referring patient to medical care which led to progression of symptoms, and influenced the patients’ social functioning, and quality of life. Early recognition and symptom improvement of this illness requires a multidisciplinary approach including patient education, awareness of physicians about different phenotypes, 28 early genetic testing, 8 and instant supplemental therapy. 39
Limitations
Biochemical investigations are required to figure out how the mutations in genes affecting CoQ activity and to examine the effect of CoQ supplementation. 20 Various biochemical methods such as high-pressure liquid chromatography (HPLC) and tandem-mass spectrometry are used in the diagnosis of primary CoQ10 deficiency. 40 Not only the technique but also the tissue of which the level of CoQ10 is measured are important for appropriate diagnosis. Biochemical analysis of patient-derived cells is also used to validate the pathogenicity of the detected variant. It is recommended to use the muscle biopsy 40 or accurately assess CSF CoQ10 status which was not possible in our study due to the time lag for the diagnosis, and patients’ disagreement of performing lumbar puncture, respectively. Moreover, assessing the status of CoQ10 and lactate after the treatment is beneficial to predict the treatment response which were not measured in this study. 23 Future studies would be beneficial for computational and in vitro analysis to clarify the pathogenicity of the detected variant.
Conclusion
We reported a case of primary CoQ10 deficiency who had rare variant of CoQ8A gene mutation (c.911C>T; [p.A304V]) with notable clinical and laboratory features including ataxia, head tremor, proximal muscle weakness, psychiatric problems and abnormal levels of urea, lactate and CoQ10. The treatment response after 2 months highlights the importance of identification, education and follow-up of patients suffering from this treatable cause of spinocerebellar ataxia.
Supplemental Material
sj-docx-1-icr-10.1177_11795476231188061 – Supplemental material for Adolescence Onset Primary Coenzyme Q10 Deficiency With Rare CoQ8A Gene Mutation: A Case Report and Review of Literature
Supplemental material, sj-docx-1-icr-10.1177_11795476231188061 for Adolescence Onset Primary Coenzyme Q10 Deficiency With Rare CoQ8A Gene Mutation: A Case Report and Review of Literature by Mahsa Hojabri, Abolfazl Gilani, Rana Irilouzadian, Habibe Nejad biglari and Roham Sarmadian in Clinical Medicine Insights: Case Reports
Footnotes
Acknowledgements
The authors would like to appreciate the patients’ family for their cooperation and patience.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
MH was the principal investigator of the study. MH, RS and AG were included in preparing the concept and design. RI and HB revisited the manuscript and critically evaluated the intellectual contents. All authors participated in preparing the final draft of the manuscript, revised the manuscript, and critically evaluated the intellectual contents. All authors have read and approved the manuscript’s content and confirmed the accuracy or integrity of any part of the work.
Ethics Approval and Consent to Participate
The informed consent and permission for the use of patient’s clinical data has been provided.
Consent for Publication
A written informed consent was obtained from the parents of the patient. All of the authors declare that confidentiality of the patient was respected.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.