
Abstract
Langerhans cell histiocytosis (LCH) is a rare disorder most commonly involving skin, bone and lung. The gastrointestinal tract (GIT) is an uncommon site of disease and only a handful of case reports exist. We present a case of a 15-year old boy with treated LCH involving the skin, bones, central nervous system (CNS) and pituitary gland. He presented with rectal bleeding and on investigation was found to have a single rectal polyp which was confirmed histologically and immunologically to be LCH. Further investigation revealed no other foci of disease.
Introduction
Langerhans cell histiocytosis is a rare disorder of uncertain aetiology.1 -3 It is caused by proliferation and dissemination of Langerhans cells, which arise from the bone marrow. LCH has been found to be due to clonal proliferation with recurrent activation mutations of BRAF V600E in up to 60% of patients.3 -5 It may be seen in any age group but the peak incidence is in children between 1 and 5 years and the reported incidence is 5 in 1 million children.2,6
Case Report
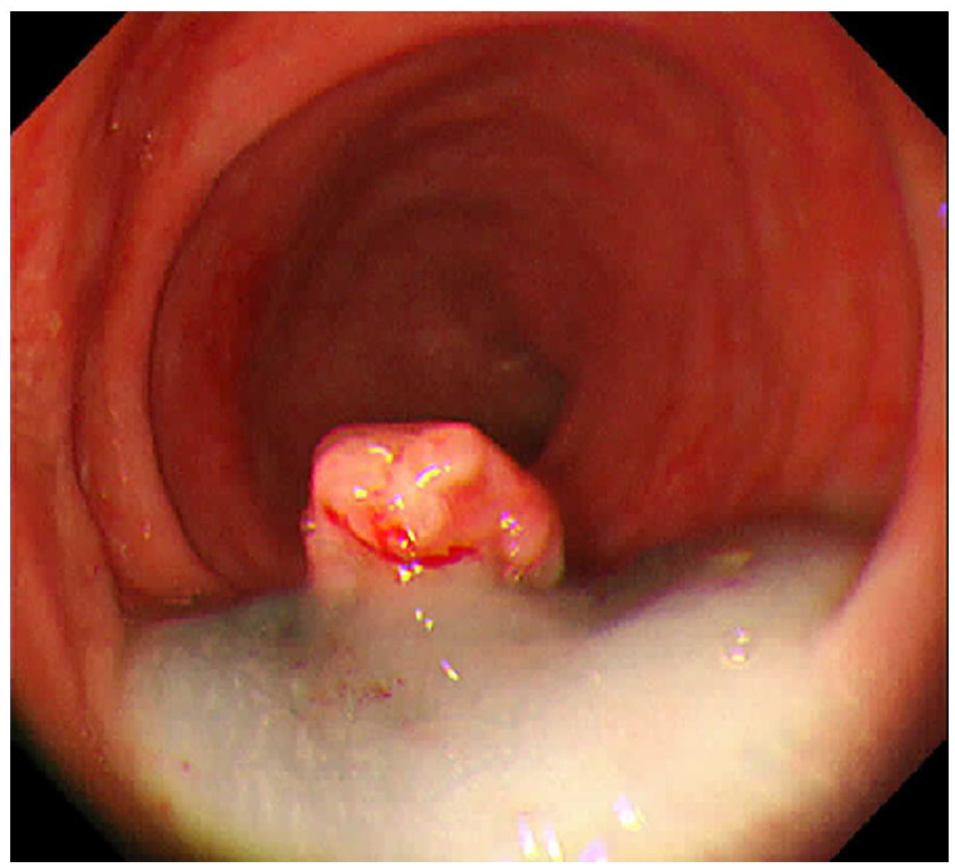
We present a case of a child who initially presented at the age of 7 years with a peri-anal skin lesion and LCH was diagnosed on his skin biopsy. Subsequent work-up including magnet resonance imaging of the brain, pelvis and skeletal survey confirmed the disease involvement of bones, CNS and pituitary gland. Rest of the imaging and bone marrow aspiration were negative. He was treated with standard first-line chemotherapy, consisting of prednisolone and vinblastine, followed by maintenance chemotherapy for 1 year. He had his first relapse with skin, skeletal and CNS involvement, 16 months after completion of treatment. Relapse was confirmed on repeat biopsy of the peri-anal lesion and positron emission tomography – computerized tomography (PET-CT). He was re-treated, with prednisolone, vincristine and cytosine-arabinoside, followed by maintenance therapy for 2 years. The patient then remained disease free for another 2 years. At the age of 15, the patient presented with painless rectal bleeding, without abdominal pain or altered bowel habit. No upper GI symptoms or weight loss was reported. He was referred to gastroenterology for further evaluation. Investigations showed a haemoglobin of 11.8 grams per deciliter (g/dl), MCV 86.3 femtoliter (fl), platelet count of 162,000/millimeter (mm3) and a normal white cell count of 6,130/mm3. Colonoscopy was performed under general anaesthesia, which was normal except for an 8 mm sessile rectal polyp, 6 cm from the anal verge, seen in Figure 1. The polyp was elevated with endoscopic mucosal resection (EMR) solution and polypectomy was performed, as seen in Figures 2 and 3. The resected polyp was submitted for histopathology. On low power (100x), sections from the resected polyps showed the typical picture of an inflammatory polyp, with dilated glands in a markedly inflamed stroma, extensive surface ulceration and granulation tissue (Figure 4). The high power view (200x), however, showed areas with histiocytic infiltrate and mixed inflammation composed predominantly of eosinophils, plasma cells and some neutrophils (Figure 5). On further examination (400x), the histiocytic infiltrate showed cells having bland nuclei with grooved membranes imparting a coffee bean-like appearance, with low mitotic activity (Figure 6). This proliferation was diffusely positive for CD1a, which is a highly sensitive and specific marker for Langerhans cell histiocytosis. S-100 immunohistochemical was also positive (Figure 7). The margins of the polyp were not involved. A PET-CT scan was subsequently carried out, to evaluate possible disease elsewhere. The PET-CT revealed no other site of fluorodeoxyglucose avid disease. The patient was tested for BRAF mutations and was found to be positive for BRAF V600E/D mutation and negative for BRAF V600K/R/M. He was then discussed in the paediatric oncology multidisciplinary meeting, where this was felt to be low risk LCH requiring only surveillance, at present. Patient is currently disease free and under follow up, since his second relapse (11 months ago).

Endoscopic view of the rectal polyp.
Elevation of the polyp with EMR solution.

After endoscopic mucosal resection of the polyp.

Polyp with dilated glands, marked inflammation, ulceration and granulation tissue (100x).
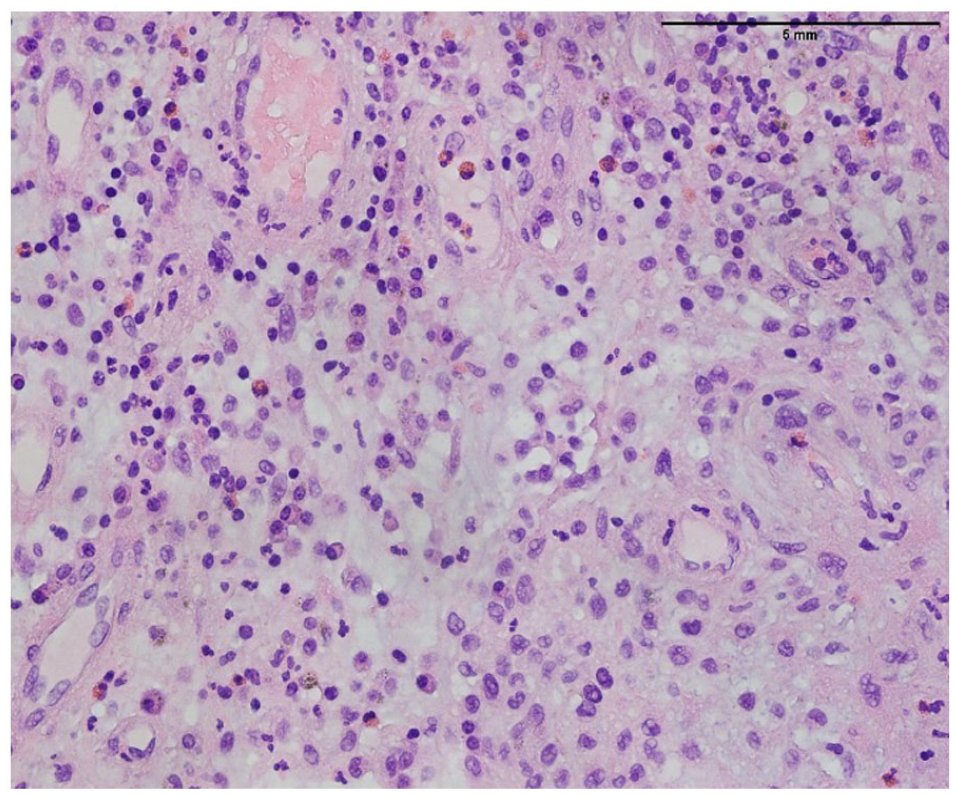
Polyp with histiocytic infiltrate and mixed inflammation (200x).

Histiocytic infiltrate with cells giving coffee bean-like appearance. Low mitotic activity (400x).

CD-Ia immunostain. Histiocytes show positive cytoplastic and membrane staining.
Discussion
LCH is characterized by single system (SS-LCH) or multisystem (MS-LCH) involvement. Possible sites of disease include the skin, skeleton, liver, endocrine glands, spleen, central nervous system (CNS), thymus and lung.2,7,8 Gastro-intestinal involvement is extremely rare, with only a few cases having been reported, worldwide, and only 1 case series being documented in the literature.1,3,6 -9 To the best of our knowledge, a total of 43 cases of paediatric intestinal LCH, had been published till 2021. 1 The literature suggests that gastro-intestinal LCH is usually associated with multisystem involvement, with a poor prognosis and a mortality rate of up to 55%.3,9 Interestingly, our patient presented with the gastro-intestinal (GI) tract as the sole site of recurrence.
Patients with GI involvement may present with variable symptoms, depending upon the area of the GI tract involved, including vomiting, abdominal pain, protein-losing enteropathy, bloody and non-bloody diarrhoea, malabsorption and failure to thrive.2 -4 In a case report by Chavananon et al, 8 the initial diagnosis of LCH in a child was missed because of the non-specific nature of the symptoms and almost all cases of GI LCH reported have had similar non-specific symptoms. 1 Our patient had a clear history of rectal bleeding and so was investigated early, with colonoscopy for this new-onset symptom on a background of previous disseminated LCH.
According to the literature, the commonly observed endoscopic findings in children with LCH are multiple erythematous and ulcerated lesions. Wang et al and Suryawanshi et al reported 2 infants and a child respectively, with such endoscopic findings in paediatric patients. 3 Adults are more likely to present with solitary, polypoid mucosal lesions.1,3,4,6,9 In our child, LCH presented with rectal polyp, adding to the rarity of this case.
Owing to the rarity of LCH with GI involvement, and the subsequent scarcity of data in the literature, no consensus has emerged as to the optimal treatment of this condition. 8 For our patient, the paediatric oncologists decided to continue surveillance, due to the single site of disease relapse, making this low-risk organ involvement, and because the resection margins of the resected rectal specimen were free of disease.
In summary, LCH of the GI tract is a rare entity and usually presents with non-specific symptoms. A low threshold for investigation in such patients ought to be maintained, so as to allow prompt diagnosis and optimal treatment, before progression to multi-system LCH, which has a much poorer prognosis.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
SMS: manuscript preparation; SB: idea and literature review; AL: pathology part was written by him; MAY: supervised and proof reading
Ethical approval
Case report has been approved by the Institutional Review Board of Shaukat Khanum Memorial Cancer Hospital and Research Centre, Lahore. IRB no. EX-28-02-22-02.
Consent statement
Written informed consent was obtained from the father of the child to publish this report in accordance with the journal’s patient consent policy.