
Abstract
Langerhans cell histiocytosis (LCH) is a rare disorder characterized by the proliferation of Langerhans cells, a type of dendritic cell essential for immune response. While LCH predominantly affects children, its manifestation in adults, especially within the gastrointestinal (GI) tract, is exceedingly rare. We present a unique case of a 56-year-old female with rare GI manifestations of LCH. The patient initially noticed pimple-like lesions around her anal orifice, which evolved into prominent protruding lesions over 3 months. Subsequent colonoscopy revealed multiple ulcers in the colorectal area, particularly concentrated in the sigmoid colon. Histopathological examination of biopsy samples, combined with immunohistochemical staining, confirmed the diagnosis of LCH. This case underscores the importance of a comprehensive diagnostic approach, especially when patients are present with atypical symptoms. The current literature suggests that such GI manifestations of LCH in adults are infrequent, making this case a valuable contribution to the understanding of LCH’s clinical spectrum.
Plain language summary
Langerhans cell histiocytosis (LCH) is an uncommon disease where certain immune cells, called Langerhans cells, grow more than they should. This disease is mostly seen in children, but it’s very rare in adults, especially affecting the digestive tract. We discuss the case of a 56-year-old woman who had unusual symptoms, including bump-like lesions around the opening of her anus that turned into larger, protruding bumps over three months. She also had multiple ulcers, mainly in the lower part of her large intestine. A detailed examination of tissue samples from her digestive tract confirmed that she had LCH. This case highlights the need for thorough medical evaluations when adults have unusual symptoms in their digestive system. The findings from this case add to the limited information we have about how LCH can appear in adults, particularly in the digestive system.
Introduction
Langerhans cell histiocytosis (LCH) is a rare and intriguing disorder, first characterized by Lichtenstein in 1953, which involves the proliferation of Langerhans cells. 1 These cells are a type of dendritic cell that plays a pivotal role in the immune response.1,2 Although LCH can manifest in individuals across all age groups, it predominantly affects children. 2 The disease can target any organ, but in children, it frequently involves the bones, skin, pituitary gland, and other organs such as the liver, spleen, hematopoietic system, and lungs. 3
The involvement of the gastrointestinal (GI) tract in LCH is particularly rare, accounting for about 2%–4% of all LCH cases. Most of these GI tract manifestations are diagnosed in children under the age of 1. 4 In adults, the occurrence of LCH in the GI tract is even rarer. When it does manifest, it often presents as isolated polyps. Ulcerative presentations, especially multiulcerative forms or lesions around the anus, are exceedingly uncommon. Globally, only a few cases with such presentations have been reported. 5
Previously, we reported part of this case. 6 Because this is a rare form of the disease, we now present this case in more detail and update some issues related to gastrointestinal LCH in adults.
Case report
A 56-year-old female observed small pimple-like lesions around her anal region 3 months before her current evaluation. Because the initial colonoscopy revealed no significant abnormalities, she was diagnosed with an anal pimple condition and subsequently referred for dermatological management. She was advised to apply Mupirocin ointment for a duration of 7–10 days and was scheduled for a follow-up and re-examination after this period. However, approximately 3 months later, the patient experienced an enlargement of the anal lesion, which became notably prominent. This was accompanied by pain during defecation and a significant weight loss of over 3 kg. Upon her return to the hospital, the lesions around the anus were observed to have evolved into pronounced protruding formations with distinct boundaries and hyperemic surfaces (Figure 1). Consequently, she was readmitted for a more comprehensive assessment. Laboratory tests indicated the presence of anemia, suggestive of microcytic hypochromic anemia, a type of anemia often associated with chronic diseases and iron-deficiency (Table 1). A subsequent colonoscopy revealed multiple ulcers in the colorectal area, with a notable concentration in the sigmoid colon. These ulcers, ranging in size from 0.3 to 1 cm, displayed raised edges, distinct boundaries, and were covered centrally by pseudomembranes (Figure 2).

(a-c) Endoscope images taken at the anal verge showed protruding perianal lesions (asterisks).
Laboratory investigation of the patient.
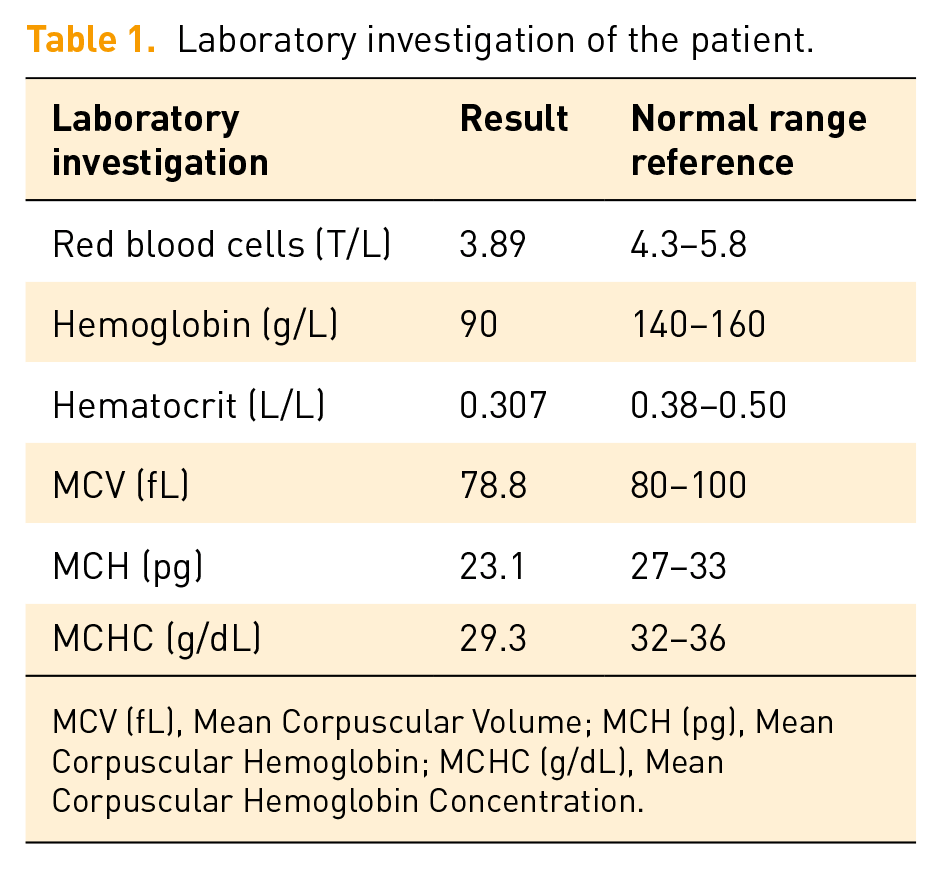
MCV (fL), Mean Corpuscular Volume; MCH (pg), Mean Corpuscular Hemoglobin; MCHC (g/dL), Mean Corpuscular Hemoglobin Concentration.

(a-c) Endoscopic images showed multiple ulcers in the colorectal area, significantly concentrated in the sigmoid colon, ranging in size from 3 to 10 mm, with raised edges, distinct boundaries and covered in the center by pseudomembrane (arrows).
The initial thought of the clinical and endoscopic physicians at that time was that this could be Crohn’s disease. Biopsies of the lesions were taken, which were then sent for histopathological examination.
Histopathological examination of the biopsy from the lesions demonstrated a proliferation of cells with reniform (kidney-shaped) nuclei, abundant eosinophilic cytoplasm, and a scattered presence of eosinophils. These cellular features on hematoxylin and eosin staining were suggestive of LCH (Figure 3). However, considering the morphological characteristics and its presentation in the GI tract, a range of differential diagnoses were contemplated. These included indeterminate cell histiocytosis (ICH), Rosai–Dorfman disease, juvenile xanthogranuloma (JXG), histiocytic sarcoma, lymphoma, melanoma, reactive langerhans cell hyperplasia (RLCH), and systemic mastocytosis. To definitively diagnose and distinguish LCH from these potential conditions, immunohistochemical staining was undertaken. The cells exhibited positivity for CD1a, S100, Cyclin D1, and langerin (CD207), thereby confirming the diagnosis of LCH (Figures 4 and 5).

Histopathological examination with hematoxylin and eosin staining of biopsied lesions taken at magnifications of (a) 40×, (b) 100×, and (c) 400× showed proliferation of cells with reniform nuclei and eosinophilic cytoplasm (circles), and scattered presence of eosinophils (arrows).

Images of immunohistochemical staining of LCH showed Langerhans cells were diffusely positive for (a) CD1a, (b) S100, and (c, d) langerin (CD207).

Images of immunohistochemical staining (a, b) showed that Langerhans cells were positive for cyclin D1.
The histopathological and immunohistochemical findings were crucial in distinguishing LCH from its mimics. The presence of Langerhans cells with their characteristic morphology on hematoxylin and eosin, combined with the immunohistochemical profile, provided a definitive diagnosis, underscoring the importance of a comprehensive diagnostic approach in such cases. To provide a comprehensive evaluation, the patient underwent a clinical skin examination and was subjected to a series of diagnostic imaging procedures, encompassing computed tomography (CT) scans, PET/CT, ultrasounds, and X-rays. However, these tests did not identify any abnormalities or lesions in other organs.
The patient was treated with low dose of cytarabine 5 days/month for a period of 12 months. Initial treatment with chemotherapy brought positive results, she had reduced signs of lesions. We have consulted and recommended testing for BRAF and MAP2K1 mutations, but the patient has not yet agreed. She is currently being monitored and evaluated regularly, and we are actively working to find a specific treatment plan for her.
Discussion
LCH is characterized by the abnormal proliferation of cells that bear a resemblance to Langerhans cells. However, it’s crucial to note that LCH originates from progenitor cells in the bone marrow and not directly from the skin’s Langerhans cells. 7 LCH was formerly referred to as histiocytosis X, until it was renamed in 1987. 8
Regarding their origin, Langerhans cells are a subtype of histiocytes. These histiocytes are essentially large white blood cells found within various tissues. The overarching category of “histiocyte” includes other cell types such as macrophages and dendritic cells. According to the World Health Organization’s classification system for hematopoietic and lymphoid tumors, histiocytic disorders are divided into three main categories: dendritic cell disorders, disorders related to macrophages, and malignant histiocytic diseases. Within this classification, LCH falls under the dendritic cell disorders, emphasizing its distinct characteristics.1,2,4
The etiology behind this cellular aggregation remains a topic of debate while some researchers argue for a reactive process, recent genetic studies lean toward a neoplastic origin. The discovery of recurrent BRAF mutations in LCH lesions, particularly the BRAF V600E mutation, has shifted the paradigm toward neoplastic etiology. This mutation activates the MAPK/ERK pathway, leading to uncontrolled cell proliferation. Other mutations, such as those in the MAP2K1 gene, have also been identified in LCH cases, further supporting the neoplastic hypothesis.1,3,5,9
LCH is predominantly diagnosed in children, but not exclusive to this age group. The involvement of the GI tract in LCH presents unique challenges. In pediatric patients, LCH often manifests with symptoms such as diarrhea, bloody stools, abdominal pain, and even failure to thrive. However, in adults, the disease frequently emerges as an incidental finding during colonoscopies, underscoring the importance of a comprehensive diagnostic approach.2,5,8,9
Within the GI tract, LCH can be present in various forms, with endoscopic findings differing based on the specific organ affected. It can manifest as polyps, ulcers, or nodular lesions. Endoscopic biopsies are pivotal for diagnosing LCH in this context. For instance, a case series by Singhi and Montgomery 5 highlighted ten instances of adult GI LCH. In the context of our patient, the identification of multiple ulcers throughout the colorectal area and a prominent anal lesion is considered a unique presentation, not commonly documented in the current literature. Therefore, diagnosing LCH is challenging. Its rare occurrence, coupled with nonspecific inflammatory patterns and a range of atypical clinical presentations, often complicates the diagnostic journey, especially in the early stages. Pathologists play a pivotal role in distinguishing these disorders. The differential diagnosis is extensive, requiring meticulous evaluation to rule out other inflammatory and neoplastic disorders.2,7,10 In the diagnostic workup of LCH, several diagnoses should be excluded, depending on the anatomic localization of the disease. 3 While the histopathological features on hematoxylin and eosin staining might hint at LCH, it’s imperative to differentiate it from other potential diagnoses, such as ICH, JXG, extranodal Rosai–Dorfman disease (RDD), histiocytic sarcoma, systemic mastocytosis, melanoma, RLCH, and lymphoma.5,7,9,10
LCH lesions are characterized by a proliferation of abnormal Langerhans cells accompanied by eosinophils, neutrophils, and occasionally lymphocytes. The abnormal Langerhans cells in LCH lesions can be distinguished from normal Langerhans cells by their larger size, pale cytoplasm, and grooved nuclei (with coffee bean- or kidney-shaped nuclei). Detection of Langerhans cell markers is essential for diagnosis. In routine practice, detection of Birbeck granules by electron microscopy has been widely replaced by detection of CD1a and CD207 expression. Immunohistochemically, these cells are positive for S-100 protein, CD1a, Cyclin D1, and CD207.3,4 Immunohistochemistry panels, including markers such as CD163, CD1a, CD207, S100, CD68, CD3, Factor VIIIa CD20, CD117, tryptase, HMB45, MelanA, and cyclin D1 can provide definitive diagnosis in LCH and differentiate it from other conditions.2,5 To highlight some key distinguishing features of LCH from its potential differential diagnoses based on immunohistochemical and histopathological characteristics, a comparison is provided in Table 2.2,4,5,8,11–14
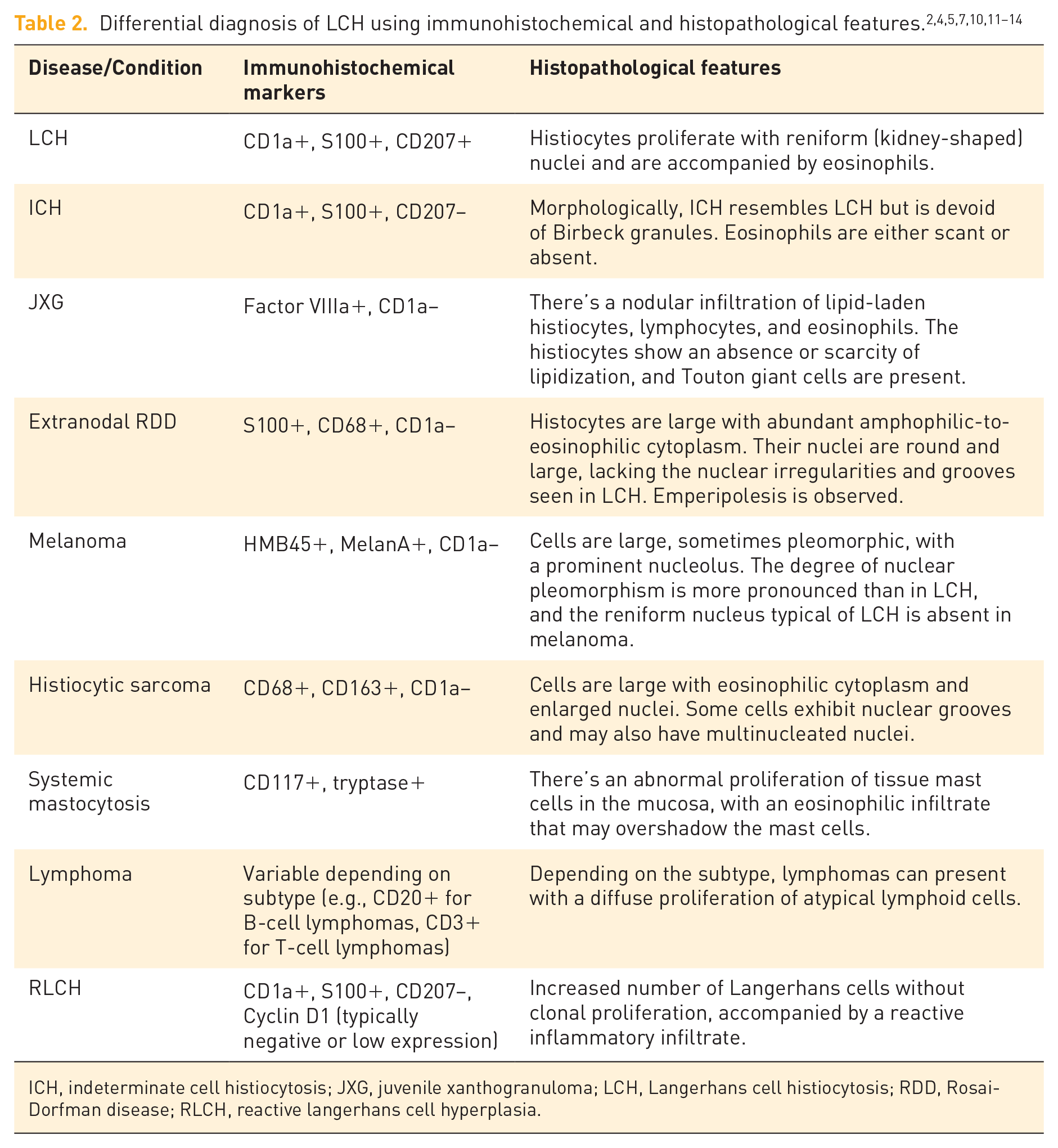
ICH, indeterminate cell histiocytosis; JXG, juvenile xanthogranuloma; LCH, Langerhans cell histiocytosis; RDD, Rosai-Dorfman disease; RLCH, reactive langerhans cell hyperplasia.
RLCH is a benign proliferation of Langerhans cells typically in response to various inflammatory or immune stimuli. While RLCH is commonly observed in the skin, lymph nodes, and other organs involved in immune response, its occurrence in the GI tract is extremely rare. Most reported cases of Langerhans cell proliferation in the GI tract are typically associated with LCH rather than RLCH. When RLCH does occur in the GI tract, it may present with nonspecific symptoms such as abdominal pain, diarrhea, or GI bleeding, which can be easily confused with other GI conditions like inflammatory bowel disease, infections, or neoplasms. Diagnosis of RLCH in the GI tract often requires endoscopic biopsies and histopathological examination, including immunohistochemical staining for markers like CD1a, S100, and CD207. Cyclin D1 is typically negative in RLCH, which helps distinguish it from LCH, where cyclin D1 expression is positive. Additionally, genetic mutations commonly found in LCH, such as BRAF V600E and MAP2K1, are not seen in RLCH. Thus, while RLCH can theoretically occur in any tissue where Langerhans cells are present, its occurrence in the GI tract is rare. Most GI tract cases with Langerhans cell proliferation are associated with LCH, a clonal and neoplastic condition, rather than RLCH, which is reactive and benign.13,14
Due to the rare nature of LCH and an incomplete understanding of the pathogenesis, treatment and chemotherapy regimens in adults are not standardized.1,4 Historically, LCH was categorized into four primary syndromes: Letterer–Siwe disease, Hand–Schüller–Christian disease, eosinophilic granuloma, and congenital self-healing reticulohistiocytosis, also known as Hashimoto–Pritzker disease.8,9,15
However, it’s important to note that many patients often exhibit symptoms that don’t align perfectly with one specific category, leading to overlapping clinical presentations. Considering this, modern clinical practice has shifted toward a more comprehensive classification of LCH. This approach categorizes LCH based on the extent of the disease (either single or multisystem) and the involvement of specific organs. The categories include unifocal LCH, single-system multifocal LCH, single-system pulmonary LCH (often associated with smoking), and multisystem LCH, with particular attention to high-risk organs such as the liver, spleen, and bone marrow.10,11,16 However, multisystem disease requires a more aggressive approach, often involving chemotherapy. The discovery of the BRAF V600E mutation has led to the exploration of targeted therapies, such as Vemurafenib, which has shown promise in refractory cases. 17 The prognosis, understandably, varies, with multifocal and multisystem diseases often leading to more challenging outcomes.9,10,16,17
Conclusion
The involvement of LCH in the GI tract is rare, representing only a small fraction of all cases. For adults, this occurrence within the GI tract is even more exceptional. Each case can exhibit a broad range of clinical manifestations. Histopathological examination, combined with immunohistochemical staining, is pivotal for confirming the diagnosis. The rarity and variability in presentation of LCH in the GI tract underscore the importance of a comprehensive diagnostic approach, especially when patients present with atypical symptoms. The current literature emphasizes the need for heightened awareness among clinicians to consider LCH as a differential diagnosis when encountering unusual GI manifestations, ensuring timely diagnosis and appropriate patient management.