
Abstract
Introduction:
Autosomal recessive non-syndromic hearing loss (ARNSHL) is a genetically heterogeneous sensorineural disorder with an approximate incidence of 1.4:1000 in neonates. Mutations in more than 60 genes including the MYO15A gene has been reported in patients affected with ARNSHL. In the present study, we report a novel MYO15A mutation identified by clinical exome sequencing and confirmed by Sanger sequencing in a consanguineous Iranian family with ARNSHL.
Case presentation:
A 22-year-old woman with congenital non-syndromic sensorineural hearing loss referred to our medical genetic center. Her parents were consanguineous with F = 1/16 (first cousin), and clinical examination of the patient exclude dysmorphic features. Sanger sequencing of GJB2 and GJB6 genes, which are the most common causes of ARNSHL, was negative. Then she underwent clinical exome sequencing.
Outcome:
We found a novel homozygote variant (c.9611_9612+8delTGGTGAGCAT) in the MYO15A gene which creates a shift in the reading frame starting at codon 3204. This variant was confirmed by Sanger sequencing in the patient and also in her parents who were heterozygous.
Discussion:
The present results suggest that the homozygous MYO15A (c.9611_9612+8delTGGTGAGCAT) variant is a pathogenic mutation and to the best of our knowledge, this mutation has not been reported in any database.
Introduction
Non-syndromic sensorineural hearing loss is the most common sensorineural disorder accounting for ~70% of hereditary hearing loss of which 80% have an autosomal recessive mode of inheritance (autosomal recessive non-syndromic hearing loss [ARNSHL]). 1 To date, more than 60 genes and more than 100 genetic loci have been identified in patients affected with ARNSHL (Hereditary Hearing Loss Homepage, http://hereditaryhearingloss.org). The genes GJB2, SLC26A4, MYO15A, OTOF, and CDH23 are most commonly implicated in ARNSHL. 2
The MYO15A mutations (NM_016239) have been reported to cause sensorineural hearing loss in human (autosomal recessive 3 [DFNB3]). 3 The DFNB3 locus (OMIM-600316) was first identified in an isolated village in Indonesia where 2% of their population was affected by hearing loss. Then, the causative role for MYO15A gene mutation was identified in DFNB3. 4 The MYO15A has 66 exons spanning 71 kb of DNA on chromosome 17p11.2. 5
The MYO15A mRNA transcript encodes a 3530 amino-acid protein (myosin XVa), which has MyTH4 (Myosin Tail Homology 4), SH3 (Src Homology 3), and PDZ domains, and FERM (4.1 protein, Ezrin, Radixin, and Moesin) motifs. Myosin XVa is a critical protein for tip localization of whirlin and differential elongation of hair-cell stereocilia and organization of actin within the hair cells of the cochlea, so myosin XVa is an important element in the normal auditory function. 6
There are several linkage analysis studies on mutations of MYO15A causing ARNSHL in consanguineous families from Pakistan, Turkey, and Iran.7-9 Mutations of MYO15A at the DFNB3 locus are the second most common cause of autosomal recessive non-syndromic deafness in Iranian population. 7 , 9 In the present study, we report a novel MYO15A mutation identified by clinical exome sequencing in a consanguineous Iranian family with ARNSHL. To the best of our knowledge, this mutation has not been reported in any database.
Materials and Methods
A 22-year-old woman with hearing loss was referred to the Medical Genetic Department, Urmia University of Medical Sciences, for detection of any possible hereditary hearing loss mutation. Her parents were consanguineous with F = 1/16 (first cousin) (Figure 1A), and clinical examination of the patient excludes dysmorphic features. Audiometric records were compatible with sensorineural hearing loss, so the non-syndromic sensorineural hearing loss was postulated.
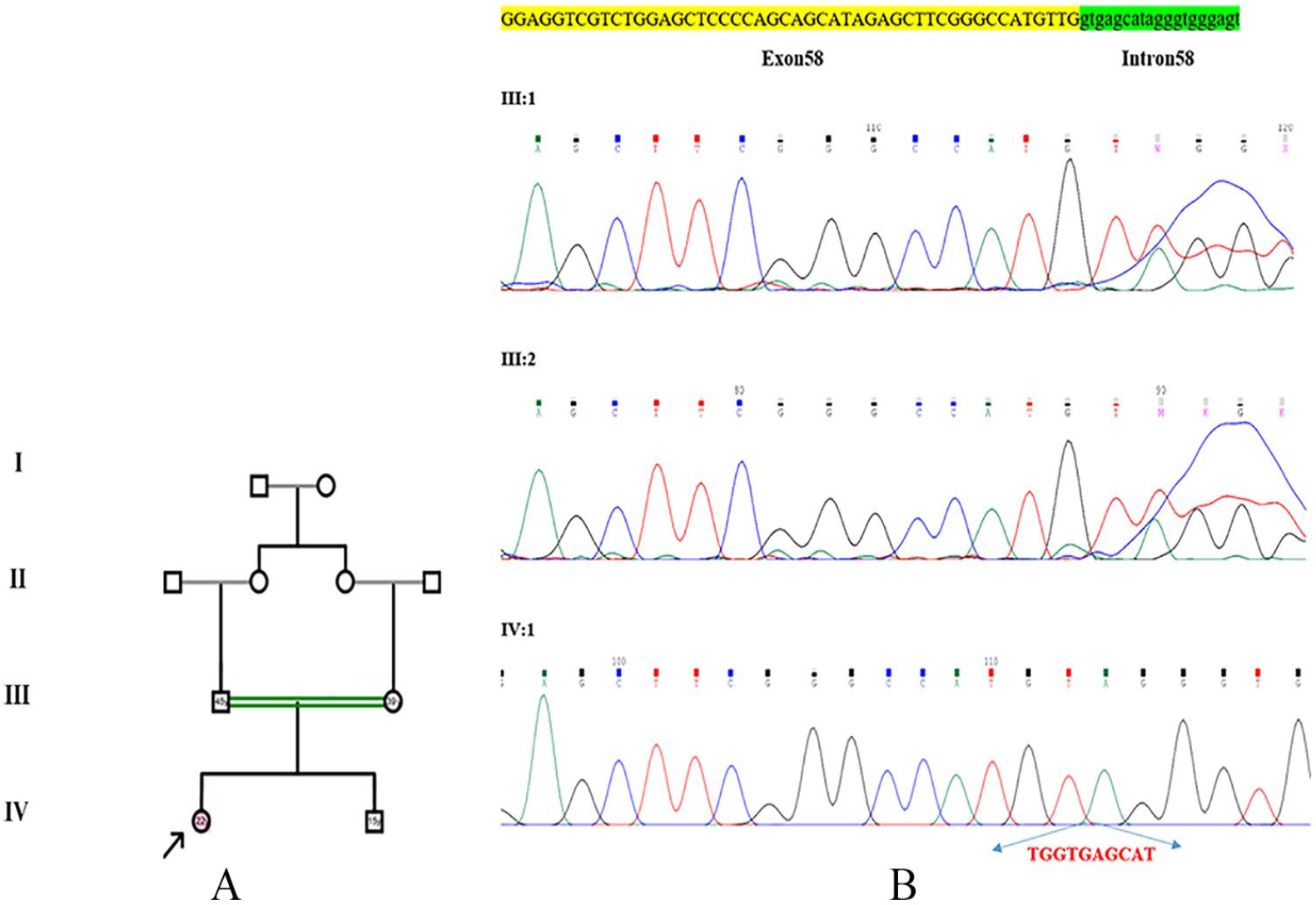
(A) The pedigree of the studied family. (B) Confirmation of novel pathogenic variant, c.9611_9612+8delTGGTGAGCAT (p.Leu3204Cysfs*17), in the MYO15A gene by Sanger sequencing.
Molecular analysis was performed after obtaining informed consent. Sanger sequencing of GJB2 and GJB6 genes which are the most common causes of ARNSHL was negative. Then, clinical exome sequencing was performed on a single proband case. Exome sequencing data were mapped to the human reference (National Center for Biotechnology Information [NCBI] build 37.1, UCSC hg19) using Burrows-Wheeler aligner (BWA-MEM, version 0.7.10). All variants were annotated by ANNOVAR and Variant Effect Predictor (VEP). Variants with a read depth > 20 and Minor allele frequency (MAF) < 0.01 in the 1000 Genome Project, dbSNP, Exome Aggregation Consortium (ExAC), and ESP-6500 were extracted for further analysis. Finally, the candidate variants were checked in the Human Gene Mutation Database (HGMD), Varsome, and Clinvar.
After preliminary detection of mutation by clinical exome sequencing, polymerase chain reaction (PCR) amplification and Sanger sequencing of the identified mutation was performed for verification of the co-segregation in the studied family. In addition, the detected mutation was searched in the healthy controls database for the same ethnic group (Iranome; http://www.iranome.ir/).
Results
A novel homozygous mutation c.9611_9612+8delTGGTGAGCAT (p.Leu3204Cysfs*17) was found in exon 58 of MYO15A in a consanguineous Iranian family with a case of non-syndromic sensorineural hearing loss (Figure 1B). This mutation creates a shift in the reading frame starting at codon 3204. The new reading frame ends in a stop codon at position 16 downstream. The deletion is near the highly conserved donor splice site of exon 58. Sanger sequencing confirmed the homozygosity of this mutation in the patient and its heterozygosity in her parents. This variant is classified as likely pathogenic (class 2) according to the recommendations of ACMG (American College of Medical Genetics and Genomics). Using the 1000 Genomes Project database and the ExAC browser, candidate variant was defined as splice-site and frame-shift mutation. The c.9611_9612+8delTGGTGAGCAT (p.Leu3204Cysfs*17) was not found in healthy controls database for the same ethnic group (Iranome; http://www.iranome.ir/). This novel mutation is predicted to disrupt the function of the myosin XVa protein, which is integral to the mechanosensory and neurosensory activity of the hair cells in the inner ear.
The alignment of the MYO15A gene from different species of human, chimpanzee, and monkey was analyzed. The result proved that this region was conservative among multiple species which is highly suggesting that these residues are important for the proper protein function.
Discussion
New sequencing technologies like clinical exome sequencing are rapidly becoming a common molecular diagnostic test for individuals with rare and heterogeneous genetic disorders. In this study, a novel mutation was identified by clinical exome sequencing within an Iranian patient with ARNSHL. Hearing loss is a highly heterogeneous disorder and the genetic causes of this disorder have been studied in Iranian populations. 10 , 11 The MYO15A mutations affect approximately 5.71% of Iranian 12 and 9.9% of Turkish ARNSHL patients. 13
Pathogenic variants in the MYO15A gene are associated with deafness type 3 which is an autosomal recessive disorder. The unconventional myosin XVa protein has an important role in the normal auditory function and other unconventional myosin proteins VI and VIIa mutations also lead to the hearing impairment. 14 Previously, several pathogenic mutations related to ARNSHL have been reported in the MYO15A gene (Table 1).
Overview of MYO15A mutations in the ARNSHL patients detected in the different populations.

Abbreviations: ARNSHL: autosomal recessive non-syndromic hearing loss; FERM, 4.1 protein, Ezrin, Radixin, and Moesin; MyTH4, Myosin Tail Homology 4; SH3, Src Homology 3.
Myosins are actin-based motor molecules with ATPase activity that drive the movement of actin filaments to facilitate organelle trafficking, cell movement, cytokinesis, signal transduction, and muscle contraction. 32 Human myosin XVa includes N-terminal domain, myosin head (motor domain), neck region, and tail domain. The motor domain contains 2 binding sites for adenosine triphosphate and actin. The neck region contains 2 light-chain binding motifs. The tail region contains 2 MyTH4 domains, 2 FERM domains, an SH3 domain, and a C-terminal PDZ-binding ligand domain. 12 Myosins highly divergent tails are required for the development and elongation of the stereocilia through the delivery of whirlin to the tips of the stereocilia. Whirlin binds to the SH3-MYTH4-FERM-domains of the myosin XVa protein and regulates actin filament elongation. Hair bundles consist of up to 300 stereocilia and are located at the apex of sensory hair cells in the cochlea and they are responsible for the mechano-electrical transduction of sound waves in the auditory system of human. 33 The architecture of the hair bundle and stereocilia length is required in the maintaining of normal hearing function. 34
In our patient, a novel homozygous deletion mutation, c.9611_9612+8del TGGTGAGCAT (p.Leu3204Cysfs*17), was identified in exon 58 of the MYO15A gene which creates a shift in the reading frame starting at codon 3204 and predicted to lie in the second MyTH4 domain of myosin XVa. Several mutations in the first and second MyTH4 domains of the myosin XVa protein have been described previously. A study in Japan revealed a homozygous mutation (p.L3138Q) in the MyTH4 domain of the myosin 15a protein in a patient with congenital severe to profound sensorineural hearing loss. 30 Also in Pakistan, sequencing of MYO15A gene in DFNB3-linked families revealed several pathogenic mutations such as a missense mutations (p.L3160F) in MyTH4 domains of the myosin XVa protein. 20 Other similar reports have been summarized in Table 1. The MyTH4 domain in the tail region is found in 1 or 2 copies and has been identified as a conserved domain in the several different unconventional myosins. Even though their specific function is not fully understood, the MyTH4 domains are predicted to be largely alpha-helical which associated with other domains like myosin head, kinesin motor, FERM, PH, SH3, and IQ. 35
Conclusions
We have identified a novel variant in the MYO15A gene in an Iranian DFNB3 family which affects second MyTH4 domain of the myosin XVa protein. The present result indicates that clinical exome sequencing enabled us to discover rare variants in the highly heterogeneous monogenic diseases like hearing loss. Further analysis of the MYO15A gene can facilitate diagnosis of congenital hearing loss in countries with a high level of consanguineous marriage.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Elinaz Akbariazar performed experiments analyzed data and wrote the paper; Ali Vahabi analyzed data; Isa Abdi Rad Designed experiments analyzed data and co-wrote the paper.
Informed Consent
Molecular analysis was performed after obtaining informed consent.