
Abstract
Background:
Pulmonary hypertension (PH) is a pulmonary vascular disease characterized by elevated pulmonary vascular pressure. Long-term PH, irrespective of its etiology, leads to increased right ventricular (RV) pressure, RV hypertrophy, and ultimately, RV failure.
Main body:
Research indicates that RV failure secondary to hypertrophy remains the primary cause of mortality in pulmonary arterial hypertension (PAH). However, the impact of PH on RV structure and function under increased overload remains incompletely understood. Several mechanisms have been proposed, including extracellular remodeling, RV hypertrophy, metabolic disturbances, inflammation, apoptosis, autophagy, endothelial-to-mesenchymal transition, neurohormonal dysregulation, capillary rarefaction, and ischemia.
Conclusions:
Studies have demonstrated the significant role of oxidative stress in the development of RV failure. Understanding the interplay among these mechanisms is crucial for the prevention and management of RV failure in patients with PH.
Introduction
PH and its effects on the right ventricle (RV) have long remained an enigmatic domain. PH encompasses a spectrum of clinical disorders characterized by elevated pressure in the pulmonary vasculature (PV), encompassing conditions such as pulmonary arterial hypertension (PAH), PH due to chronic pulmonary veno-occlusive disease, PH secondary to left heart disease, PH arising from lung disease and hypoxia, chronic thromboembolic pulmonary hypertension (CTEPH), and PH stemming from multifactorial mechanisms. 1 These disorders are unified by the common feature of increased pulmonary vascular resistance (PVR) attributable to excessive pulmonary vasoconstriction and aberrant vascular remodeling. 2 Consequently, these events collectively impose heightened afterload on the right RV, precipitating subsequent changes.
PH instigates progressive afterload-induced remodeling of the RV, characterized by augmented contractility (to uphold RV-arterial coupling), diastolic stiffening, RV dilation, and eventual progression to right heart failure. 3 The prognostic significance of RV function in PH is well recognized. 4 The stroke volume-to-end-systolic volume ratio has demonstrated prognostic utility in PAH patients, with a ratio below 0.5 indicative of RV volume augmentation to sustain stroke volume. 5 However, the gold standard for assessing RV function is the end-systolic-to-arterial elastance (Ees/Ea) or coupling ratio. 6 RV-arterial decoupling is posited as the underlying pathomechanism of RV maladaptation and failure in PH. Multi-beat Ees/Ea has been identified as an independent prognostic factor, with a threshold of 0.7. 7 Despite these advancements, the precise effects of PH on the RV and its structural consequences remain inadequately elucidated. This review aims to consolidate recent insights into the ramifications of PH on the RV, commencing with a succinct overview of RV alterations in PH and subsequently delving into specific details of these changes.
Right Ventricle Changes in Pulmonary Hypertension
In the early stages of PH, the RV maintains stroke volume by augmenting wall thickness (hypertrophy) and enhancing muscle contractility, termed homeometric adaptation. However, with the progression of PH, the inefficiency of RV systolic function leads to RV dilation, the only effective response to increased afterload, termed heterometric adaptation. This increase in RV end-diastolic volume corresponds to a parallel increase in stroke volume, thereby ensuring RV output. 3 Impaired ventricular relaxation, cardiac wall stiffness, and subsequent elevation in cardiac filling pressures are associated with diastolic dysfunction. 8 In advanced stages of PH, diastolic dysfunction and further RV dilation contribute to leftward septal bowing, subsequently impeding left ventricular (LV) function and exacerbating systolic dysfunction (manifested as reduced stroke volume). 3 These structural changes impair RV function and contribute to increased morbidity and mortality. Notably, RV failure secondary to hypertrophy remains the primary cause of death in PAH.9,10
Extracellular remodeling
Matrix metalloproteinases and tissue inhibitors of metalloproteinases
RV morphological changes, characterized by extensive structural and extracellular matrix (ECM) remodeling, ultimately culminate in right heart failure and mortality. 9 Aberrant accumulation of ECM proteins and significant cardiac fibrosis are hallmarks of RV remodeling. 10 Studies have demonstrated alterations in ECM protein composition within the RV matrix in patients with PH, potentially associated with increased morbidity and mortality. 11
The ECM plays a critical role in regulating myocardial function (contraction and relaxation) by maintaining ventricular geometry, integrity of cardiac structure, and elasticity of heart tissue. 9 Furthermore, the ECM governs the functions of cardiac cells, including myocytes, pericytes, fibroblasts, endothelial cells, and smooth muscle cells, influencing processes such as cell migration, adhesion, proliferation, differentiation, and survival, thus preserving normal cellular homeostasis. 9
ECM homeostasis relies on a controlled balance between matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs), regulating ECM-associated proteins during cardiac remodeling. 9 Dysregulation of MMPs and TIMPs can lead to cardiac fibrosis. 12 There are four types of TIMPs, each with distinct roles: TIMP-1 promotes fibrosis 13 ; TIMP-2 contributes to cell proliferation 14 ; TIMP-3 induces apoptosis 15 ; and TIMP-4, predominantly expressed in cardiac tissue, induces apoptosis in transformed cells without affecting normal cells. 13 Elevated levels of MMPs and TIMPs have been observed in patients with PH compared to healthy individuals, indicating increased collagen turnover and ECM remodeling. 16 Experimental models of myocardial hypertrophy and failure have also revealed upregulation of MMPs and altered collagen expression, contributing to adverse cardiac remodeling. 17
Deposition of ECM contributes to RV stiffness and development of heart failure. 18 Among ECM-associated proteins, MMPs are crucial contributors to PH-induced RV remodeling, primarily expressed by fibroblasts and cardiomyocytes. 17 Additionally, ADAMTS proteinases, another group of extracellular metalloproteinases, play a significant role in degrading ECM-affiliated proteins, proteoglycans, and procollagens, potentially affecting cardiomyocyte-ECM integrity and function during RV failure progression.19,20 Loss of ECM integrity may significantly contribute to ventricular remodeling and cardiac dysfunction. 9
Increased oxidative stress can lead to the generation of nitro-tyrosine residues in TIMPs, liberating active MMPs. Imbalance in the MMP-to-TIMP ratio results in interstitial fibrosis. Pulmonary artery constriction is associated with increased formation of reactive oxygen species (ROS) and RV failure. Studies have shown that folic acid can mitigate ROS generation, maintain the MMP-to-TIMP ratio, and reduce interstitial fibrosis in a mouse model of pulmonary artery constriction by increasing TIMP-4 levels and mitigating increased MMP-2, −9, and −13 levels (Figure 1). 21
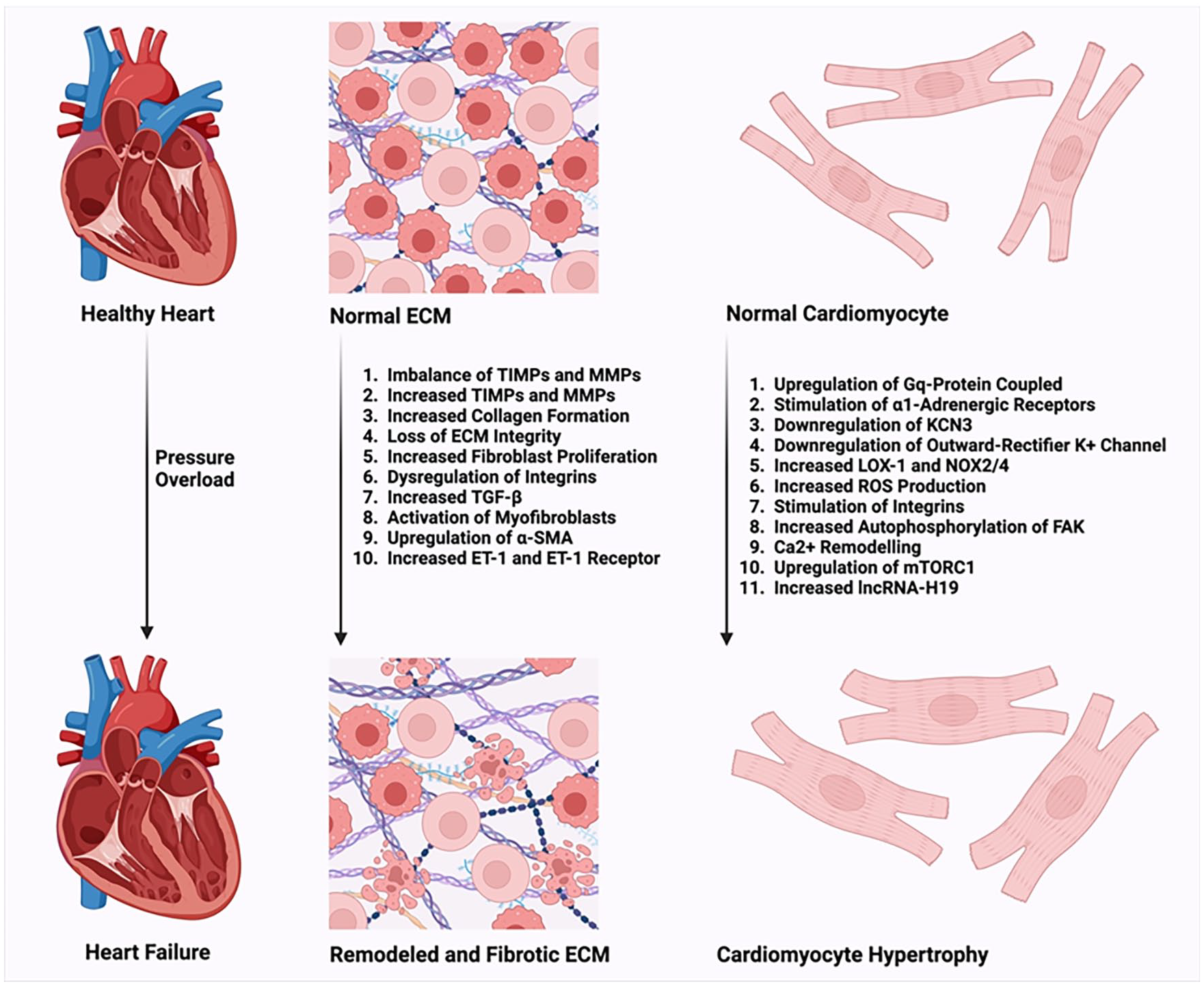
RV morphological changes encompass ECM remodeling, ultimately culminating in right heart failure. The ECM regulates the functions of various cardiac cell types, including myocytes, pericytes, fibroblasts, endothelial cells, and smooth muscle cells. Moreover, it plays a crucial role in processes such as cell migration, adhesion, proliferation, differentiation, and survival, thereby contributing to the preservation of normal cellular homeostasis. Additionally, the RV responds to increased afterload by undergoing hypertrophy.
Collagen production
Every cardiac disease, regardless of its etiology, involves variable degrees of fibrosis, representing a complex interplay of adaptive and maladaptive changes. Pressure overload, such as in PH, leads to enhanced collagen formation initially, aimed at maintaining normal ventricular shape and withstanding high pressures. However, over time, adaptive collagen accumulation transitions to maladaptive alterations in the collagen network structure and ECM integrity, ultimately contributing to cardiac dysfunction and disease progression. 9
Pressure overload induces increased collagen synthesis, particularly collagen I secretion, leading to collagen deposition.22,23 Late-stage increase in insoluble collagen due to pulmonary artery banding has been associated with RV diastolic dysfunction. 24 Increased collagen deposition is accompanied by high collagen turnover driven by MMP and TIMP activation, exacerbating fibrosis. This high collagen turnover stimulates abnormal ECM protein formation and fibrosis. Sustained pressure overload leads to fibroblast proliferation and increased collagen production, while myofibroblasts isolated from ventricles exposed to pressure overload display increased migration, proliferation, and enhanced contractility.25,26
These alterations are associated with dysregulation of integrin expression. Integrins connect fibroblast contractile filaments to ECM-bound transforming growth factor-β (TGF-β), releasing TGF-β upon fibroblast contraction or ECM stretch, which activates myofibroblasts, upregulates α-smooth muscle actin (α-SMA), and enhances collagen production. 27 Similarly, cardiomyocytes respond to mechanical stress by producing growth-stimulating factors, including TGF-β and angiotensin-II (Ang-II; Figure 1).28,29
Galectin-3 (Gal-3) emerges as a novel biomarker of left cardiac remodeling and exerts influence over various pathophysiological processes, including oxidative stress, inflammation, fibrosis, and immunity. In patients with PAH, Gal-3 levels are elevated. Investigations on MCT-induced PAH rat models revealed an upregulation of Gal-3 and NOX-4 in the right ventricle. Areas abundant in Gal-3 were found to co-localize with collagen III deposition. Furthermore, the administration of Gal-3 recombinant protein stimulated the proliferation, differentiation, collagen deposition, and NOX-4 expression of cardiac fibroblasts. Notably, the profibrotic effects induced by TGF-β1 on cardiac fibroblasts were partially mediated by Gal-3. Gal-3 facilitated TGF-β1-induced cardiac fibrosis through interaction with NOX-4 and NOX-4-derived oxidative stress. These stimulatory effects were effectively hindered through Gal-3 knockdown. 30
Furthermore, emerging evidence suggests that endothelin-1 (ET-1) may mediate RV fibrosis and dysfunction. Upregulation of endothelin and endothelin type A receptor in the RV of patients with PAH and experimental RV pressure overload models has been demonstrated. 31 ET in the RV may stimulate fibroblast proliferation and promote synthesis of ECM proteins. 32 Research indicates that hemodynamic overload in adult pigs with aortocaval fistula leads to heightened ET-1 levels, triggering an ensuing augmentation in myocyte diameter and length, as well as collagen deposition in the RV. 33 Moreover, ET receptor blockade has been associated with attenuation of RV fibrosis in various models of PH.34-36
Right ventricle hypertrophy
As previously mentioned, in the initial stages of pulmonary hypertension (PH), the right ventricle (RV) responds to increased afterload through hypertrophy. Various mechanisms have been proposed, including pressure overload, neurohormonal factors, 37 gene regulation, 38 mitochondrial remodeling, 39 signaling pathways, 40 and ROS production. 41
Regulatory factors and molecular pathways
A hallmark of RV dysfunction/hypertrophy associated with PH is the downregulation of KCNK3, encoding an outward-rectifier K+ channel. Lambert et al 38 demonstrated reduced KCNK3 function in RV cardiomyocytes during the development of RV hypertrophy (RVH) in rat models of PH, and chronic inhibition of KCNK3 in rats was associated with RVH. Additionally, several factors, such as downstream signaling of all Gq-protein-coupled receptors including endothelin-1 (ET-1), tyrosine kinase activity, and platelet-derived growth factor (PDGF) receptor, can modulate KCNK3 channel functionality.42,43 Furthermore, 1A-adrenergic receptor stimulation inhibits KCNK3. 44
Long non-coding RNAs (lncRNAs), transcripts longer than 200 nucleotides, have been implicated in the development of various diseases and cellular processes. 45 Omura et al 46 revealed that lncRNA H19 is upregulated in the RV of patients with PAH and correlates with RVH and RV fibrosis. Silencing H19 was associated with limitation of RVH and RV fibrosis, preserving RV function in rat models without affecting pulmonary vascular remodeling.
The mTORC1 pathway serves as a master regulator of lipid, protein, and nucleotide synthesis, contributing to cell growth and the hypertrophic response. 47 Upregulation of mTORC1 has been observed in the SU5416/hypoxia rat model of pulmonary hypertension (PH), associated with cardiomyocyte and right ventricular (RV) hypertrophy. However, there was only a marginal increase in mTORC2/Akt signaling that did not elevate with PH. Moreover, inhibition of mTOR kinase reversed cardiomyocyte hypertrophy and RV remodeling. 48
It appears that HIF1 and HIF2 regulate RV remodeling in chronic alveolar hypoxia (CH), with HIF1 playing a protective role rather than driving hypertrophy. Deletion of HIF-1α augments CH-induced hypertrophy. 37 HIF-1α deletion contributes to the upregulation of G-coupled protein receptor signaling, tyrosine and protein phosphorylation, and signal transducer and activator of transcription, which can augment hypertrophy. 37 Smith et al 37 showed that deletion of HIF-1α in vascular smooth muscle, not cardiomyocytes, attenuates CH-induced PH. HIF-1α in vascular smooth muscle cells contributes to PH and pulmonary vascular remodeling in chronic hypoxia. However, the inaction of HIF-1 in smooth muscle does not affect RV remodeling, suggesting that cardiac hypertrophy is not directly linked to the increase in pulmonary artery pressure. 49
Pressure overload is transmitted to cardiomyocytes via integrins, which activate intracellular messengers such as the autophosphorylation of focal adhesion kinase (FAK). Increased phosphorylated FAK has been observed in RVH in mouse models of PH. 17 Stimulation of integrins and FAK contributes to a hypertrophic response in cardiomyocytes (Figure 1). 50
Oxidative stress
Oxidative stress has been implicated in cardiomyocyte hypertrophy. Research by Redout et al 41 demonstrated that antioxidant therapy in monocrotaline (MCT)-induced PAH attenuated RV hypertrophy and associated changes in mRNA expression (such as deiodinase type 3 and myosin heavy chain-β). Additionally, antioxidant therapy reduced RV oxidative stress, proapoptotic signaling, and prevented interstitial fibrosis. Mechanical stretch of isolated cardiomyocytes increases ROS production, which acts as a downstream effector of various hypertrophic stimuli.51,52
The lectin-like oxidized low-density lipoprotein receptor (LOX)-1, an endothelial receptor of oxidized low-density lipoprotein, plays a role in ROS generation. LOX-1 promotes superoxide production via NADPH oxidase (NOX) and subsequently upregulates ROS-dependent molecules such as the activation of NF-κB. 53 Under hypoxic conditions, LOX-1 transgenic mice exhibited greater right ventricle systolic pressure (RVSP) and RV hypertrophy compared to wild-type mice. Hypoxia also enhanced ROS formation and nitro-tyrosine expression in LOX-1 transgenic mice, supporting the aforementioned pathological changes. 54 Zhu et al 55 reported that hypoxia contributes to RVH via the upregulation of LOX-1 and subsequent increase in NOX2/4, triggering ROS generation. Moreover, NOX-2 plays a role in the development of Ang-II-induced cardiac hypertrophy. 56 Hypoxia-induced hypertrophy in rats’ hearts and H9C2 cell lines (cardiomyocytes) via upregulation of NOX2 and NOX4, leading to increased oxidative stress. Knockdown of LOX-1 attenuated H9C2 hypertrophy by significantly reducing oxidative stress, accompanied by a decrease in NOX2 and NOX4 protein expression. 55 Additionally, pressure overload leads to upregulation of NOX4, primarily residing in mitochondria. 57
Metabolism derangements of right ventricle
Aerobic glycolysis
PH is associated with increased RV oxygen consumption due to tachycardia, increased wall thickness, and RV hypertrophy. This increased demand for oxygen should ideally be met by either increasing myocardial perfusion or enhancing the fraction of oxygen extracted from blood. However, limited myocardial blood flow in PH poses a risk of ischemia and hibernation. 58 Possible factors contributing to reduced blood flow to the RV in PH include a reduction in systolic and diastolic pressure gradient between the aorta and right ventricle, as well as capillary rarefaction in the hypertrophic RV. 59
Moreover, PH is associated with metabolic changes in the RV, particularly at the mitochondrial level, which affect RV efficiency. 60 Under basal conditions, the RV generates energy predominantly through fatty acid oxidation (FAO; ~70%), with a smaller contribution from glycolysis or lactate utilization. 61 However, a phenomenon known as the Warburg effect (aerobic glycolysis) has been observed in pressure-overloaded RV cardiomyocytes, where glycolysis becomes the predominant energy production pathway, and mitochondrial FAO is inhibited.62,63
Pyruvate dehydrogenase kinase (PDK) plays a crucial role in mediating this shift toward aerobic glycolysis in PAH. Increased levels of PDK isoforms have been observed in PAH, and inhibition of PDK has been associated with improved RV contractility in PAH patients (Figure 2). 64
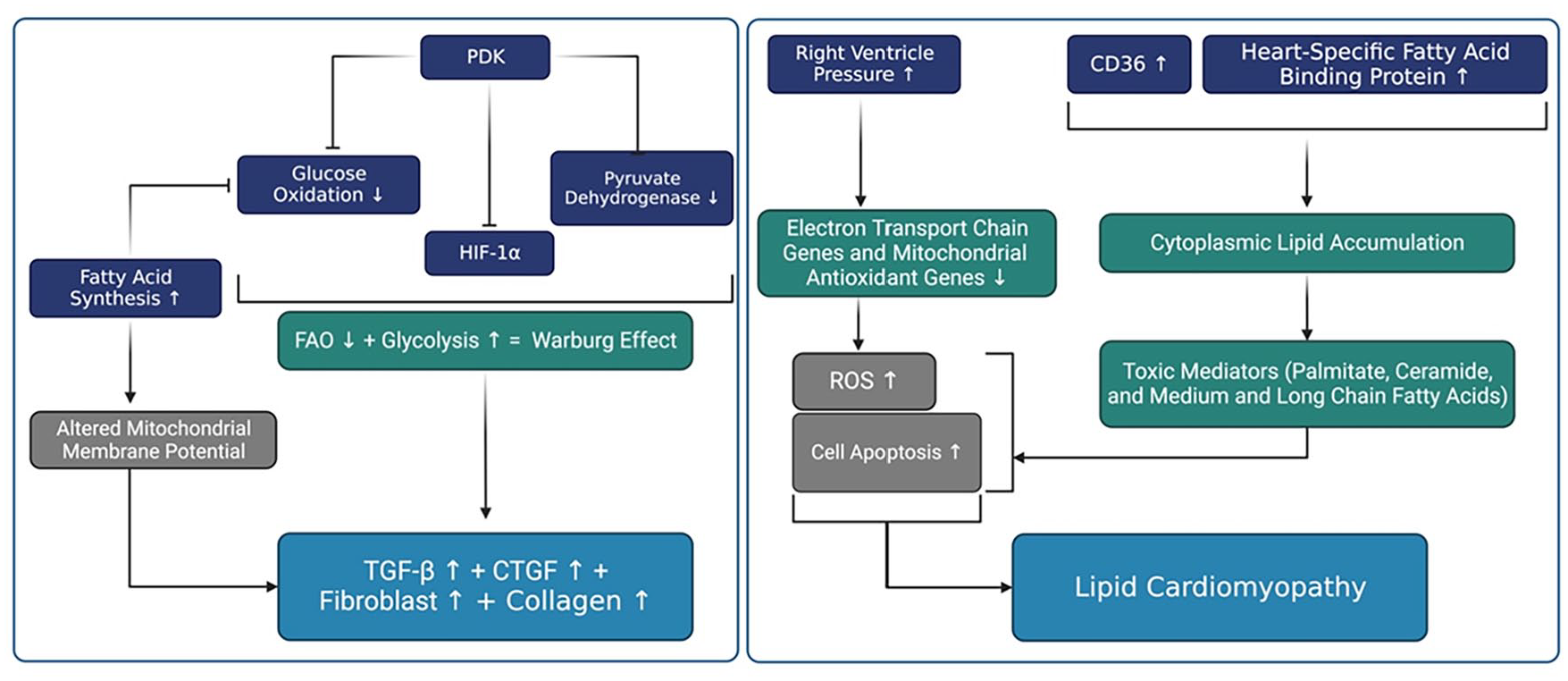
Is associated with increased RV oxygen consumption due to factors such as tachycardia, increased wall thickness, and RV hypertrophy. Metabolic changes occur in the RV in PH, particularly at the mitochondrial level, which impacts RV efficiency. Studies have demonstrated the presence of the Warburg effect (aerobic glycolysis) in pressure-overloaded RV cardiomyocytes, wherein glycolysis becomes the predominant mode of energy production over fatty acid oxidation (FAO). Decreased mitochondrial FAO contributes to lipid accumulation in the cytoplasm of cardiomyocytes. This lipid accumulation leads to the production of toxic mediators, including palmitate, ceramide, and medium and long-chain fatty acids, which can contribute to the development of lipotoxic cardiomyopathy through excessive generation of reactive oxygen species and cell apoptosis.
Additionally, studies have demonstrated that dichloroacetate, a PDK inhibitor, reduces phospho-pyruvate dehydrogenase expression, leading to reduced RV fibrosis and hypertrophy, and enhanced RV function. 65
Reducing the activity of PDK isoforms 1 and 3 restores mitochondrial superoxide and hydrogen peroxide production, which inactivate the hypoxia-inducible factor (HIF)-1α. Redox-mediated inactivation of HIF-1α downregulates transforming growth factor-β (TGF-β) and connective tissue growth factor (CTGF), leading to reduced fibroblast proliferation and collagen production. 65
As previously mentioned, these events are associated with increased RV fibrosis and dysfunction.
Furthermore, downregulation of metabolic master regulators, including PPARγ, PPARγ coactivator (PGC1α), and PPARα, has been observed in the RV under conditions of pressure overload.66,67 In line with this, pioglitazone, a PPAR agonist, has been shown to reverse PAH and prevent right heart failure in mouse models, primarily through FAO induction and the upregulation of key genes such as CPT1b and FABP4. 68
Significant decreases in FAO and an increase in cytoplasmic lipid accumulation have been demonstrated in the RV myocardium of patients with PAH.62,69 CD36, responsible for 60% of fatty acid uptake to cells, is upregulated in the RV of mice with BMPR2 mutation-related PAH. 69 Additionally, increased levels of heart-specific fatty acid-binding protein have been identified as an independent predictor of adverse events in patients with chronic thromboembolic PH. 70
This dysregulation of metabolism contributes to lipid accumulation in the cytoplasm of cardiomyocytes, leading to the production of toxic mediators such as palmitate, ceramide, and medium and long-chain fatty acids, which can contribute to lipotoxic cardiomyopathy through excessive reactive oxygen species production and cell apoptosis (Figure 2). 71 Inhibition of fatty acid synthesis has been shown to improve glucose oxidation, mitochondrial membrane potential, ATP production, and decrease hypertrophy, inflammation, apoptosis, and autophagy in hypoxic cardiomyocytes. Moreover, inhibition of fatty acid synthesis in monocrotaline (MCT)-treated rats decreased RV hypertrophy and improved cardiac function. 72
Mitochondrial dysregulation and oxidative stress
Mitochondrial dysregulation has been implicated in the progression of RV dysfunction. Studies have shown increased variability in mitochondrial size, decreased mean mitochondrial diameter, mitochondrial disarray, and an increased number of autophagosomes near mitochondria in mouse models of PAH. 68 Additionally, RV pressure overload has been associated with decreased expression of electron transport chain genes and mitochondrial antioxidant genes, as well as increased expression of oxidant stress markers, contributing to decreased energy generation. 73
Mitochondrial dysfunction may also play a role in insulin resistance, which has been implicated in pathologic pulmonary vascular remodeling in pre-clinical models of PAH. 74 Serum insulin levels have been found to inversely correlate with right ventricular function and lung volumes in patients from the general population free of cardiovascular disease. 75 Additionally, it seems that RV may be more prone than LV to changes in insulin-related signaling. 76 However, more studies are needed to elucidate the role of insulin-related signaling in RV dysfunction in patients with PH.
Increased levels of ROS generated by NOXs and mitochondria have been reported in the RV of patients and rats with PAH.77,78 Antimycin, a mitochondrial complex III inhibitor, and apocynin, a Nox-2 inhibitor, have been shown to decrease ROS levels in the RV, suggesting that elevated neurohormonal factors and pressure overload activate NOX-2 and mitochondrial electron transport chain/NOX-4, leading to ROS generation. 79
Glucose-6-phosphate dehydrogenase (G6PD) is a major source of NADPH, which is a substrate for NADPH oxidase (NOX) in the heart. Altered NADPH signaling in the failing heart of Sugen5416/hypoxia-induced PH has been associated with increased oxidative stress, metabolic maladaptation, and autophagy, contributing to RV remodeling and hypertrophy. Reduction in G6PD-derived NADPH, by inhibition of G6PD, has been associated with decreased oxidative stress, increased glucose oxidation and acetyl-CoA, reduced autophagy, and consequently reduced PH and RV hypertrophy. Moreover, reduced G6PD-derived NADPH has been associated with improved myocardial contractility and decreased cardiomyocyte L-type Ca2+ currents. 79
Upregulation of oxidative stress-related genes has been observed in the RV 24 hours after pulmonary artery constriction, including p47phox, NOX2, and Rho kinase (ROCK2). Additionally, dominant-negative Rho kinase mice did not show the presence of oxidative stress species, indicating the involvement of oxidative stress in RV remodeling. 80 Dehydroepiandrosterone (DHEA), a steroid that decreases NADP levels in the RV, downregulates Rho kinase, resulting in the reduction of active forms of STAT3 and NFATc3, which prevents RV remodeling. The antioxidant activity of DHEA reduces RVSP and preserves contractility in PH. 81
Calcium remodeling
RV hypertrophy (RVH) is associated with calcium (Ca2+) remodeling. Decreased rates of Ca2+ sequestration contribute to prolonged isovolumic contraction in MCT-treated rats with compensated RVH, while enhanced Ca2+ sequestration and mobilization during the contraction/relaxation cycle have been observed in the early stages of RVH.82,83 RV myocytes from MCT-treated rats with RVH and heart failure exhibit smaller and slower Ca2+ transients with decreased sarcoplasmic reticulum density. However, conflicting findings have been reported regarding cell shortening and an increase in Ca2+ transients amplitude associated with a higher sarcoplasmic reticulum Ca2+ load in failing myocytes from MCT-induced PH rats.83-85
Store-Operated Ca2+ Entry (SOCE) is known to participate in Ca2+ influx in cardiac cells. SOCE comprises three essential components: the regulatory element (STIM1-2), and two Store-Operated Ca2+ channels, namely, Orai (Orai1-3) and TRPCs (Transient Receptor Potential Canonical 1-7). STIM1 serves as an endoplasmic/sarcoplasmic reticulum Ca2+ sensor and initiates TRCPs- and/or Orai-mediated SOCE in response to the stored Ca2+ content. 86 Sabourin et al 87 demonstrated that STIM1/Orai1/TRPC1/C4-dependent Ca2+ current plays a role in Ca2+ remodeling in pulmonary hypertension (PH)-induced right ventricular hypertrophy (RVH).
Immune system activation
The activation of the immune system is a well-known characteristic of chronic heart failure. 88 Elevated release of pro-inflammatory cytokines, such as tumor necrosis factor (TNF)-α and interleukin (IL)-6, correlates with the advancement of heart failure and portends a poorer prognosis.89,90 Both systemic and local stressors can foster an inflammatory milieu within the overloaded right ventricle (RV). Key local stressors encompass ischemia and mechanical stress, while systemic influences involve activated immune cells, heightened levels of proinflammatory cytokines, and neurohormonal activation. 91
It has been demonstrated that treatment with umbilical cord blood-mesenchymal stem cells in rats with monocrotaline-induced pulmonary hypertension significantly improves RV function and decreases medial wall thickness, perivascular fibrosis, vascular cell proliferation, as well as reduces levels of recruitment of adaptive and innate immune cells and related inflammatory cytokines. 92
Inflammatory response
Various immune cells, including mast cells, dendritic cells, macrophages, T cells, and B cells, have been identified in perivascular fibrotic regions, contributing to adverse outcomes in PAH.93-95 T cells stimulate B cells, prompting antibody secretion and the activation of macrophages into classically activated (M1) and alternatively activated (M2) macrophages, promoting Th1 and Th2 responses, respectively. 96 Activated M1 macrophages drive inflammation and phagocytosis, eliminating dysfunctional and deceased cardiomyocytes, while activated M2 macrophages resolve inflammation and sustain structural integrity through collagen deposition. 97
Notably, macrophage-derived inflammatory cytokines, such as IL-6, IL-1β, and TNF-α, correlate with cardiac remodeling, encompassing hypertrophy, fibrosis, and apoptosis. 98 CD4+ helper T cells produce mediators that impede fibroblasts and macrophages, exacerbating lung fibrosis progression. 99 CD4+ T cells have also been implicated in influencing cardiac remodeling and scarring. 100 Targeting T cells with antibodies has shown promise in reversing cardiac fibrosis in systemic sclerosis patients and a mouse model of inflammation-associated fibrosis. 101
von Haehling et al 102 compared the levels of pro-inflammatory cytokines, including TNF-α, its soluble receptors 1 and 2 (sTNFR1 and 2), IL-10, IL-6, high-sensitivity C-reactive protein, and NT-proBNP, between CHF patients and healthy subjects. They observed similar levels of immune activation, indicated by elevated levels of pro-inflammatory cytokines, in patients with isolated RV dysfunction due to chronic thromboembolic pulmonary hypertension (CTEPH) and patients with CHF and left ventricular dysfunction. IL-1β and IL-18 levels are higher in PAH patients compared to healthy subjects and are associated with worse outcomes. 103 Moreover, TNF-α and interleukin family cytokines, including IL-2, 4, 6, 8, 10, and IL-12p70, are indicative of RV failure in PAH patients.103,104 Cytokines such as TNF-α and IL-1 increase cardiac inducible nitric oxide synthase (iNOS), further contributing to mitochondrial and calcium dysfunction and disruption of the neuro-adrenergic system.105-107 Elevated levels of inflammatory cytokines are associated with increased mortality and decreased survival chances in PAH patients.103,108
Furthermore, Trankle et al 109 assessed the efficacy of anakinra (IL-1 receptor antagonist) in seven PAH patients with RV failure. They demonstrated that anakinra significantly improved heart failure symptoms and reduced inflammation. NLRP3 serves as an intracellular danger sensor, oligomerizing upon activation and contributing to the formation of inflammasomes. This process leads to the maturation and release of IL-1β. Guo et al 110 showed that inhibition of NLRP3 attenuates lipopolysaccharide-induced RV failure in rats with monocrotaline-induced PAH. Moreover, their findings suggest that inhibiting NLRP3 is associated with suppressed inflammation and increased activation of M1 macrophages.
Cardiac fibroblasts secrete extracellular matrix upon activation by various inflammatory signals and stressors to prevent ventricular rupture. 111 In instances of cardiac injury, fibroblasts differentiate into myofibroblasts. Additionally, myeloid cells, wound macrophages, and epidermal and endothelial cells can serve as other origins for myofibroblasts.30,112 Myofibroblasts reconstruct cardiac tissue through extracellular production and signaling transition to neighboring cardiomyocytes and stromal cells. 11 Increasing numbers of activated and secretory cardiac myofibroblasts are major drivers of cardiac fibrosis, characterized by excessive fibrillary collagen Iα deposition and maladaptive RV remodeling. 113
Oxidative stress and immune dysregulation
In vivo and in vitro studies have revealed that mechanical stress directly initiates cardiac inflammation by locally producing reactive oxygen species (ROS) and subsequently activating NF-κB.114-116 Oxidative stress and PH-induced cytokines upregulate NF-κB expression. NF-κB binds to the nitric oxide synthase 2 (NOS-2) promoter, increasing NOS-2 transcription. The RV utilizes nitric oxide (NO) to counteract increased afterload by promoting pulmonary vasodilation. RV pressure overload induces NOS-2 in cardiac fibroblasts, accompanied by increased formation of reactive oxidants. Induction of NOS-2 and increased formation of reactive oxidants contribute to collagen formation, resulting in cardiac fibrosis and dysfunction. 117 Chronic hypoxia and prolonged exposure to NO have been shown to contribute to complete RV dysfunction. Therapeutic hypercapnia (10% CO2) blocks the interleukin (IL)-1α pathway, inhibiting NOS-2, and subsequently normalizing RV hypertrophy. 118
ROS carbonylates annexin A1 protein, leading to its degradation by proteasomes in the RV. Annexin A1, primarily expressed by leukocytes, is a principal dysregulated gene in decompensated rat hearts, inhibiting endothelin-1-associated inflammatory cytokine secretion. 119 Annexin A1 knockout in mice was associated with proinflammatory gene expression, fibrotic gene expression, and collagen I deposition in the left ventricle after myocardial infarction. 120 PH reduces the interaction between annexin A1 and CBF/NY-F, associated with activation of the GATA4 gene transcription. GATA4 is a hypertrophy regulator in the RV.121,122 Moreover, dysregulation of annexin A1 was identified in RV transcriptomic analysis of monocrotaline-induced pulmonary hypertension in Sprague-Dawley rats. Additionally, this analysis showed that CYP2E1, a protective gene against oxidative stress, is downregulated in pulmonary hypertension. 119
Apoptosis and autophagy
Apoptosis
Chronic pressure overload contributes to increased cardiomyocyte size and force of contraction, a phenomenon known as cardiac hypertrophy. This hypertrophy is associated with quantitative and qualitative changes in the myocardium, including angiogenesis, which is a significant alteration. However, pressure overload eventually leads to a mismatch between the size of cardiomyocytes and capillary density, resulting in myocardial hypoxia. RV ischemia has been observed in PAH patients due to the loss of RV microvessels and increased oxygen demand. 123
The combined use of a vascular endothelial growth factor (VEGF) receptor inhibitor and hypoxia in rats has demonstrated similar properties to those observed in human patients with severe PAH. This model exhibited signs of RV failure, including cardiomyocyte apoptosis and cardiac fibrosis. 124 Campian et al 125 investigated the role of apoptosis in monocrotaline-induced PH in rats and found that the progression of RV failure is associated with an early increase in RV apoptosis. Moreover, acute increases in RV afterload have been shown to upregulate apoptosis inducers such as p53 and Bax, leading to subsequent increases in cardiomyocyte apoptosis. 126 P53 binds to the CBF/NF-Y transcription factor and negatively regulates GATA4. 127 Edmondson et al demonstrated increased apoptosis, downregulation of the anti-apoptotic protein BCL-xL, downregulation of GATA4 (a transcriptional regulator for BCL-xL), and upregulation of p53 (a negative regulator of GATA4 gene transcription) in a rat model of PAH. Additionally, PAH-induced RV apoptosis and fibrosis were attenuated in p53 knockout rats. 128 Li et al 129 revealed increased RV cardiomyocyte apoptosis in MCT-treated rats due to decreased Bcl-2 and upregulation of Bax, Bok, and active caspase-3. Prolonged left-to-right shunt-induced PAH in piglets increases the pro-apoptotic Bax/Bcl-2 ratio and activates caspase-3 (the final executioner of apoptosis), associated with increased apoptosis and RV failure. 130
As previously mentioned, dichloroacetate improves mitochondrial function. Similarly, Sun et al demonstrated that administration of dichloroacetate in rats with MCT-induced PH was associated with improved cardiac function and reversed RV remodeling. They showed that dichloroacetate downregulates pyruvate dehydrogenase kinase (PDK) and upregulates pyruvate dehydrogenase, contributing to restored mitochondrial function and subsequent reductions in apoptosis, hypertrophy, fibrosis, and capillary rarefaction. Dichloroacetate administration upregulated Bcl-2 and downregulated Bax expression. Moreover, dichloroacetate attenuated cytochrome c leakage from mitochondria and decreased the levels of caspase-3, caspase-9, and poly (ADP-ribose) polymerase (PARP). All these alterations are associated with decreased mitochondria-induced apoptosis. 131
Furthermore, increased apoptosis in hypertrophied RV in a pulmonary artery banding (PAB) model in rats has been reported, associated with upregulation of the Bax protein and caspase-3. However, the expression of Bcl-2 was not altered in the experimental group. 132
PAR formation depolarizes and decreases mitochondrial membrane potential. 133 Upregulation of mitochondrial PAR in the RV of MCT-treated rats has been shown to contribute to the opening of mitochondrial transition pores and the release of death factors from mitochondria, including AIF and cytochrome c, to the nucleus and cytosol, respectively.134,135 Cytochrome c, a regulator of apoptosis, is released from mitochondria and binds to pro-apoptotic proteins to create an apoptosome, which in turn activates caspase-3. 135 Kaur et al demonstrated that inhibition of PARP prevents mitochondrial dysfunction and the release of death factors from mitochondria (AIF and cytochrome c) in RV cardiomyocytes. Moreover, it decreases apoptosis by reducing the number of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and caspase-3 activity in RV cardiomyocytes. 136 Additionally, it has been demonstrated that overstretching is associated with increased expression of FAS on cardiomyocytes. 137 FAS-FAS ligand (FASL) signaling promotes apoptosis through FAS-associated protein with death domain (FADD), which in turn activates caspase-8. 138 Furthermore, inhibition of apoptosis signal-regulating kinase 1 (ASK1) reduces the remodeling of the RV and pulmonary vasculature in rats with PH. GS444217, an ASK1 inhibitor, reduces fibrosis and improves cardiac function. Additionally, GS-444217 decreases the phosphorylation of p38 and c-Jun N-terminal kinase (JNK) and reduces cardiac fibroblast activation (Figure 3). 139

Apoptosis and autophagy. Increased right ventricle pressure is associated with elevated apoptosis, characterized by the downregulation of BCL-xL (an anti-apoptotic protein), downregulation of GATA4 (a transcriptional regulator for BCL-xL), and upregulation of p53 (a negative regulator of GATA4 gene transcription). Additionally, decreased Bcl-2 levels and upregulation of Bax, Bok, and active caspase-3 have been observed. Furthermore, right ventricle failure is linked with heightened autophagy, as evidenced by increased levels of autophagy markers such as p62 and LC3. AMPK phosphorylates and activates proteins of the tuberous sclerosis complex 2 (TSC2), which subsequently deactivates mTOR, thereby stimulating autophagy.
Autophagy
Moreover, RVF has been shown to be associated with increased autophagy (p62 and LC3 markers) in a murine model of pulmonary artery constriction. 21 P62 forms complexes with various proteins and plays a role in regulating NF-κB. 140 Moreover, p62 facilitates the packing and delivery of aggregated and misfolded proteins for disposal through the autophagolysosomal degradation pathway. 141 LC3 is a well-known binding partner of p62 and is enriched on intracellular membranes during autophagy. 142 Additionally, Qipshidze et al 21 showed that folic acid decreases ROS generation and improves RV function via decreasing the autophagy and mitophagy. Levels of the autophagy marker p62 and the mitophagy marker LC3A/B increased in pulmonary artery-constricted mice, and folic acid abolished these effects. ROS inhibit autophagy-related gene 4 (Atg4) activity by oxidizing an essential cysteine residue, which can block the cleavage of phosphatidylethanolamine (PE) from PE-conjugated LC3 and promote autophagy. 143
Deng et al showed that MCT-induced PH and subsequent RV hypertrophy are associated with sustained activation of autophagy. They observed upregulation of LC3 mRNA and protein expression, increased p62 levels, and accumulation of autophagosomes in the RV of rats with MCT-induced PH. Notably, pro-autophagy signaling pathways exhibited activation in an RV-remodeling stage-dependent manner. Specifically, phosphorylated-AMPK (adenosine monophosphate-activated protein kinase)-α was upregulated, while phosphorylated-mTOR (mammalian target of rapamycin) was downregulated during RV hypertrophy and dilation stages. Conversely, BNIP3 (Bcl2-interacting protein 3) and beclin-1 expression remained low during these stages, but were reversed during RV decompensation and failure. 144
BNIP3 plays a pivotal role in regulating cardiomyocyte mitochondrial function and survival during hypoxia/ischemia injury. Its activation leads to mitochondrial perturbations, including opening of the mitochondrial transition pore, loss of mitochondrial membrane potential, and cell death. However, despite alterations in BNIP3 expression, HIF-1α (upstream molecular target of BNIP3) levels remained unchanged.144,145 BNIP3-deficient mice exhibited preserved systolic performance and reduced cardiac dilation post-infarction, whereas BNIP3 overexpression was associated with impaired systolic function and increased cardiac dilation due to heightened mitochondrial apoptosis and mitophagy. Additionally, BNIP3 can directly induce apoptosis by acting on Bcl-2.146,147
The initiation of autophagy requires an initial membrane nucleation step, facilitated by the class-III phosphoinositide 3 kinase (PI3K) complex, including Beclin-1, which is a downstream molecular target of BNIP3. Beclin-1 deficiency leads to decreased autophagic activity, preserved cardiac function, and partial inhibition of cardiomyocyte death after aortic banding, while Beclin-1 overexpression is linked to increased cardiac remodeling (Figure 3).148,149
Zhai et al demonstrated that NF-κB significantly activates autophagy, leading to downregulation of RND3 expression and promotion of MCT-induced PAH. They also revealed that AMPK activation reduces autophagy by inhibiting NF-κB in MCT-induced PAH. RND3, a member of the small Rho GTPase family, regulates various cellular processes, including migration, proliferation, differentiation, and apoptosis.150,151 In contrast, Matsui et al reported that low or depleted ATP levels or an increase in cytosolic AMP levels lead to AMPK activation. AMPK, in turn, phosphorylates and activates proteins of the tuberous sclerosis complex 2 (TSC2) and deactivates mTOR, thus stimulating autophagy. Inhibition of AMPK is associated with decreased autophagy and reduced cardiomyocyte survival, suggesting a protective role for AMPK-dependent autophagy during glucose deprivation. 152 In summary, the role of AMPK in autophagy regulation appears paradoxical, with increased autophagy observed in animal models of PH. Further research is warranted to elucidate the precise role of AMPK in autophagy under conditions of pulmonary hypertension.
Endothelial to mesenchymal transition
During endothelial-to-mesenchymal transition (EndMT), endothelial cells acquire markers of mesenchymal cells and express α-SMA. 153 Recent studies have shown that EndMT and epithelial-to-mesenchymal transition (EMT) are implicated in cardiovascular disease and fibrosis. Following cardiac injury, EndMT and EMT contribute to the conversion of endothelial and epithelial cells into myofibroblasts, leading to increased collagen deposition, fibrosis, and cardiac dysfunction. 154 Moreover, EndMT has been implicated in myocardial fibrosis and diastolic dysfunction in rodent models of pressure overload.154,155
Park et al conducted a comparative RNA-seq analysis between Sugen/hypoxia- and MCT-induced PAH in a rat model. They found similar gene enrichment in EMT, metabolism, and inflammation. Additionally, histological examination of human RVs from patients with PAH and RV failure revealed significant EndMT and RV fibrosis, along with upregulation of cellular communication network factor 2 (CCN2), a top gene implicated in EMT and EndMT, particularly in perivascular areas compared to normal RV. 156 CCN2, also known as CTGF, is a matricellular protein induced following cardiac injury and associated with tissue fibrosis. 157 Similarly, Williams et al 158 showed elevated CTGF expression in PAH-induced RV failure samples compared with nonfailing RV.
Shi et al 159 revealed that MCT-induced PH in rats is associated with deposition of α-SMA and collagen in the pulmonary artery adventitia. They also noted an upregulation of mesenchymal markers including N-cadherin, α-SMA, and vimentin, coupled with a downregulation of endothelial markers such as CD31 and VE-cadherin. Additionally, they observed an upregulation of promoting genes, namely SNAI1 (Snail) and SNAI2 (Slug).
Reduced bone morphogenetic protein receptor type 2 (BMPR2) is associated with increased EndMT. 160 Moreover, NF-κB promotes EndMT through the regulation of BMPR2. 161 Shi et al 159 also demonstrated that MCT-induced PAH is associated with decreased BMPR2 levels and increased levels of the NF-κB p65 subunit. However, their evaluation did not extend to EndMT in the RV. Histological examination revealed increased interstitial fibrosis, alongside elevated protein levels of α-SMA and lysyl oxidase (LOX), a crucial enzyme in pulmonary fibrosis development, in the RV. They further showed that administration of Baicalein, a natural flavonoid, reverses these alterations and reduces EndMT. Meanwhile, Baicalein attenuates the right ventricular systolic pressure (RVSP) and ameliorates RV fibrosis. 159
Zeisberg et al demonstrated in a mouse model of pressure overload that cardiac fibrosis is linked to an increase in fibroblasts originating from endothelial cells through EndMT. Additionally, they revealed that bone morphogenetic protein 7 (BMP-7) preserves the endothelial phenotype, while transforming growth factor beta 1 (TGF-β1) induces EndMT in a Smad-dependent manner. 155 It has been elucidated that TGF-β1 promotes EndMT, at least in part, via microRNA-21 through the phosphatase and tensin homolog/Akt pathway (Figure 4). 162

Endothelial to mesenchymal transition. During endothelial-to-mesenchymal transition (EndMT), endothelial cells acquire characteristics of mesenchymal cells. This process involves the upregulation of mesenchymal markers such as N-cadherin, α-smooth muscle actin (α-SMA), and vimentin, while simultaneously downregulating endothelial markers including CD31 and VE-cadherin.
Neurohormonal dysregulation
Targeting the neurohormonal system has long been a foundational therapeutic approach in LV-related diseases. However, there is comparatively less understanding regarding the role of the neurohormonal system in RV pathological conditions, particularly in the context of RV overload, as observed in pulmonary PH.
Under physiological conditions, β1- and β2-adrenergic receptors (ARs) enhance cardiac contractility, rate, and frequency of relaxation through the activation of G-protein-coupled receptors. This activation subsequently triggers adenylyl cyclase/cAMP/protein kinase A signaling, leading to the phosphorylation of L-type calcium channels and ryanodine receptors, thus regulating calcium handling and the Na+/K+-ATPase pump. 163 Atrial and ventricular stretch stimulate the sympathetic nervous system activation. 164 Prolonged sympathetic nervous system activation has been demonstrated to result in the downregulation and desensitization of α1- and β1-receptors in hypertrophied RVs in both PAH patients and rodent models of PH. 165 Reduced β-ARs and signaling contribute to RV dysfunction in at least two ways. Firstly, it leads to reduced contractile reserve, resulting in atrioventricular uncoupling, RV dilation, decreased exercise capacity, and diminished survival.166-168 Secondly, it impairs diastolic function due to increased cardiac stiffness. Protein kinase A, a downstream target for β-ARs, regulates cardiomyocyte stiffness via the phosphorylation of titin. 169 Attenuated β-ARs signaling contributes to reduced protein kinase A-mediated phosphorylation of titin, thereby increasing the stiffness of RV cardiomyocytes. 23 Prolonged adrenergic activation has been associated with cardiac apoptosis, hypertrophy, fibrosis, and reduced capillary density in experimental models of PAH.170,171
Furthermore, PH is associated with increased activity of the renin-angiotensin-aldosterone system (RAAS). Patients with progressive PAH, as opposed to stable patients, exhibit higher levels of Ang-I and Ang-II, contributing to disease progression and mortality. 172 Similarly, patients with PAH demonstrate elevated plasma aldosterone concentrations compared to patients with unexplained dyspnea without PAH. 173 Moreover, aldosterone levels correlate negatively with cardiac output and positively with pulmonary vascular resistance. 173 However, a study involving 125 PAH patients revealed that aldosterone levels did not correlate with the 6-minute walk test, cardiac output, or survival. 174 Sustained RAAS activation leads to adverse effects on heart including, conduction system disturbances, fibrosis, and cardiomyocyte hypertrophy. 175 Unlike the lungs, studies have reported that Ang-II type 1 receptor (AT1R) is downregulated, while ACE expression and Ang-II formation are increased in the RVs of PAH patients. 176 It has been demonstrated in MCT- and Sugen/hypoxia-induced PH mouse models that renal denervation reduces RV cardiomyocyte cross-sectional area, with a greater effect observed in Sugen-hypoxia models than in MCT-induced PH models. Additionally, renal denervation decreases RV fibrosis in Sugen-hypoxia models, further highlighting the role of renal denervation in reducing mineralocorticoid receptors and subsequently improving RV diastolic stiffness by reducing fibrosis. 177
Increased activity of the neurohormonal system may trigger inflammation in the lungs and heart in PAH. 91 Elevated sympathetic nervous system activity increases cardiac inflammation and upregulates TNF-α and IL-1β. 178 Bilateral sympathectomy has been shown to reduce oxidative stress and prevent ventricular remodeling and functional decline. 179 Similarly, bisoprolol, a β-blocker, has been demonstrated to improve cardiac function and reduce cardiac inflammation in experimental PH rats. 180 Both Ang-II and cytokines upregulate ET-1 expression, and ET-1 promotes the expression of cardiac pro-inflammatory cytokines and activates the neurohormonal system. 91 Moreover, Ang-II receptor blockade reduces cardiac fibrosis in diabetic rat models by attenuating EndMT. 181 Similarly, losartan has shown cardiac protective effects in hypertensive rats by reducing fibrosis and EndMT through inhibiting TGF-β/Smad signaling. 182
Capillary rarefaction and ischemia
Increased ventricular wall stress is associated with elevated oxygen demand, necessitating a corresponding increase in vascularization to meet metabolic needs adequately. Insufficient blood supply contributes to inadequate oxygen delivery to the RV, potentially leading to the transition from RVH to right heart failure (RHF). 3 Additionally, heightened wall stress during systole compresses intramural arteries, resulting in biphasic blood flow, which limits perfusion during diastole. PH has been linked with progressive impairments in right coronary artery flow and reduced RV perfusion. 59 Furthermore, decreased RV microvascular density and consequent RV ischemia may contribute to maladaptive remodeling. 183
Mild RV interstitial fibrosis can also impede capillary-cardiomyocyte contact surface, associated with the upregulation of genes (cardiomyocyte myosin-heavy chain isoform switch gene) reflecting impaired cardiomyocyte contractile function and hypoxia. 184 Studies have demonstrated reduced capillary density in RV samples from animal models of PH and patients with PAH. RV biopsies from PAH patients with RV failure (RVF) have shown downregulation of vasculoprotective and angiogenic genes, including apelin, angiotensin-I, vascular endothelial growth factor (VEGF), and miR-126, as well as reduced microvascular density compared to patients with compensated RVH.185,186
Research by Graham et al 187 suggests that decreased capillary density results from the failure to maintain the optimal ratio of capillaries to myocyte cross-sectional area due to a lack of proportional increase in microvasculature. Cardiomyocyte hypertrophy without adequate angiogenesis creates a ventricular chamber too large to sustain adequate tissue oxygenation, leading to significant ischemia and subsequent RV dysfunction. 187
PH in both humans and animals has been associated with increased RV capillary growth, which positively correlates with RVH. Studies by Kassa et al have revealed that mice with deletion or inhibition of prolyl hydroxylase (PHD2), HIF-1α, or 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) exhibit impaired RV adaptation during hypoxic PH. PHD2 regulates HIFs and transcriptionally activates proangiogenic genes such as PFKFB3. 188 Moreover, impaired mitochondrial function and subsequent increase in reactive oxygen species (ROS) production can inhibit HIF-1α and suppress angiogenesis. 189
Conclusion
Chronic PH culminates in RV hypertrophy, remodeling, and eventual failure. Various mechanisms, including ECM alterations, apoptosis, autophagy, metabolic disorders, EndoMT, and vascular abnormalities, have been implicated in the pathogenesis of RV dysfunction and failure. As discussed earlier, excessive oxidative stress, a component of these mechanisms, plays a pivotal role in the initiation and progression of RV failure associated with PH. Consequently, targeting RV oxidative stress presents a promising avenue for the development of novel therapeutic strategies for managing RV failure in the context of PH.
Footnotes
Acknowledgements
None.
Authors’ Contributions
S.P.E conceptualized the study. All of the authors performed the literature search and wrote the draft. E.H and S.P.E edited the article. M.N and S.P.E have the co-first authorship and contributed equally to this work.
Data Availability Statement
Data sharing is not applicable for this narrative review. No data were generated and analyzed for this article.
Declaration Of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval and Consent to Participate
Not applicable.
Consent for publication
Not applicable