
Abstract
Background:
Congenital heart disease (CHD) is the most common congenital malformation, it is frequently found as an isolated defect, and the etiology is not completely understood. Although most of the cases have multifactorial causes, they can also be secondary to chromosomal abnormalities, monogenic diseases, microduplications or microdeletions, among others. Copy number variations (CNVs) at 22q11.2 are associated with a variety of symptoms including CHD, thymic aplasia, and developmental and behavioral manifestations. We tested CNVs in the 22q11.2 chromosomal region by MLPA in a cohort of Colombian patients with isolated CHD to establish the frequency of these CNVs in the cohort.
Methods:
CNVs analysis of 22q11.2 by MLPA were performed in 32 patients with apparently isolate CHD during the neonatal period. Participants were enrolled from different hospitals in Bogotá, and they underwent a clinical assessment by a cardiologist and a clinical geneticist.
Results:
CNVs in the 22q11.2 chromosomal region were found in 7 patients (21.9%). The typical deletion was found in 6 patients (18.75%) and atypical 1.5 Mb duplication was found in 1 patient (3.1%).
Conclusions:
CNVs in 22q11.2 is a common finding in patients presenting with isolated congenital cardiac disease, therefore these patients should be tested early despite the absence of other clinical manifestations. MLPA is a very useful molecular method and provides an accurate diagnosis.
Keywords
Background
Congenital heart disease (CHD) is the most common congenital malformation and is an important cause of childhood morbidity and mortality.1,2 The incidence of severe and moderate CHD ranges from 2.5 to 3 per 1000 live births worldwide, 3 and the prevalence is reported to be continuously increasing to a maximum of 9.4 per 1000 in 2017 4 when also the mild defects are included. In Colombia, according to data from ECLAMC (in Spanish: Estudio Colaborativo Latinoamericano de Malformaciones Congénitas), the incidence is 1.2 per 1000 live births. 5 However, it is suspected that it may have a higher incidence than reported, but its inherent clinical variability may be associated with underdiagnosis.4,6,7
The etiology of CHD is still not completely understood; however, the advances in molecular techniques had shown a strong role for genetics factors.1,8 Determining the specific cause for the CHD should be based according to the presentation of the defect. The clinician should determine if the defect is part of a genetic or chromosomal syndrome according to additional findings or if it could be the consequence of maternal chronic diseases or teratogens exposure or finally if it is an isolated defect with or without a family history.9,10 The latter being the most common presentation.1,3
The chromosomic region 22q11.2 contains several paralogous low copy repeats (LCR22) that predispose to the genomic reorganization and makes it very unstable, therefore susceptible to deletions and duplications. The misalignment of low copy repeats during nonallelic homologous recombination leads to deletion or duplication of the 22q11.2 region, resulting in 22q11.2 deletion syndrome (Mendelian inheritance in man [MIM] number 611867)11,12 and 22q11.2 duplication syndrome (MIM number 608363). 13
22q11.2 deletion syndrome is a common genetic disorder with an autosomal dominant inheritance pattern, but with most of the cases presenting de novo, 14 it is characterized by velopharyngeal insufficiency, thymus aplasia leading to immunodeficiency, hypoparathyroidism, and CHD,15,16 mainly resulting from conotruncus, embryonic aortic arches, and ventricular septum. 16
Although both deletions and duplications are expected to occur in equal proportions as a result of reciprocal LCR-mediated events, fewer duplications of 22q11.2 have been described 17 most of them with an identical 3 Mb duplication. 22q11.2 microduplication syndrome has an extremely variable phenotype, ranging from multiple defects to mild learning difficulties, sharing features with 22q11.2 deletion syndrome, including heart defects, urogenital abnormalities, velopharyngeal insufficiency, and with some individuals being essentially normal. 18
In this study, we tested CNVs in the 22q11.2 chromosomal region by MLPA in a cohort of Colombian patients with isolated apparently isolated CHD during the neonatal period in order to establish the frequency of these CNVs in the cohort. Most of our patients were in the newborn period and during this period the diagnosis can be challenging because many extracardiac features will not became apparent until later in life. 19
Methods
Subjects
The study included 32 children referred to the Human Genetics Institute at Javeriana University in Bogotá, Colombia. They were referred from different hospitals in the city and from the ECLAMC program at San Ignacio Hospital from October 2012 to May 2013. Criteria of referral was: Individuals with the CHD associated with 22q11.2 deletion syndrome: Ventricular septal defect (CIE-10 Q21.0), atrial septal defect (CIE-10 Q21.1) with or without patent ductus arteriosus (CIE-10 Q250), aortic coarctation (CIE-10 Q251) tetralogy of Fallot (CIE-10 Q21.3), transposition of great vessels (CIE-10 Q20.3), double outlet right ventricle (CIE-10 Q20.1), pulmonary atresia (CIE-10 Q25.5), and overriding aorta (CIE-10 Q25.4). Exclusion criteria included cleft lip, cleft palate, mental retardation, and any other major malformation. Javeriana University’s Ethics Committee approved the study protocol, and we obtained the written informed consent from the parents of the participants.
Clinical evaluation
Before enrolment in the study, subjects underwent an echocardiogram to confirm the CHD and an evaluation by a clinical geneticist for the assessment of facial features to ruled out any major malformation. Blood calcium levels were not evaluated at the first stage.
DNA extraction
DNA from venous blood was extracted using a commercial kit (QIAamp DNA Mini Kit QIAGEN). All DNA samples were quantified by NanoDrop ND1000 Spectrophotometer (NanoDrop Technologies) and stored at −20°C.
MLPA analysis
The SALSA MLPA P250-B1 DiGeorge kit (MRC-Holland) was used to identify copy number variations (CNVs) in the 22q11.2 region according to the manufacturer’s instructions (www.mrc-holland.com). We used 1 positive and 2 negative controls. PCR amplification products were separated by capillary electrophoresis using ABI-Prism 310 Genetic Analyzer (Applied Biosystems), and the data were analyzed using the Coffalyzer analysis software (MRC-Holland).
Results
Phenotypic analysis
About 32 patients presenting with isolated CHD were included in the study. Fourteen were female (34.3%) and 21 (65.6%) male, with ages ranging from 1 day to 13 years. Figure 1 shows the type of CHD found in the 32 patients.

Cardiac anomalies found in the 32 subjects.
Molecular analyses
The MLPA test was performed on all patients admitted to the Genetics Institute. CNVs in the 22q11.2 chromosomal region was found in 7 patients (21.8%) using the MLPA P250-B1 DiGeorge kit. Six patients (18.75%) showed the typical 3 Mb deletion spanning molecular probes CLTCL1 to LZTR1 (LCRA-D) (rsa 22q11.2(P250-B1 DiGeorge LCRA-D)x1) (Figure 2a) and in 1 patient (3.1%) was found the atypical 1.5 Mb duplication spanning molecular probes CLTCL1 to DGCR8 (LCRA-B) (rsa 22q11.2(P250-B1 DiGeorge LCRA-B) × 3) (Figure 2b).
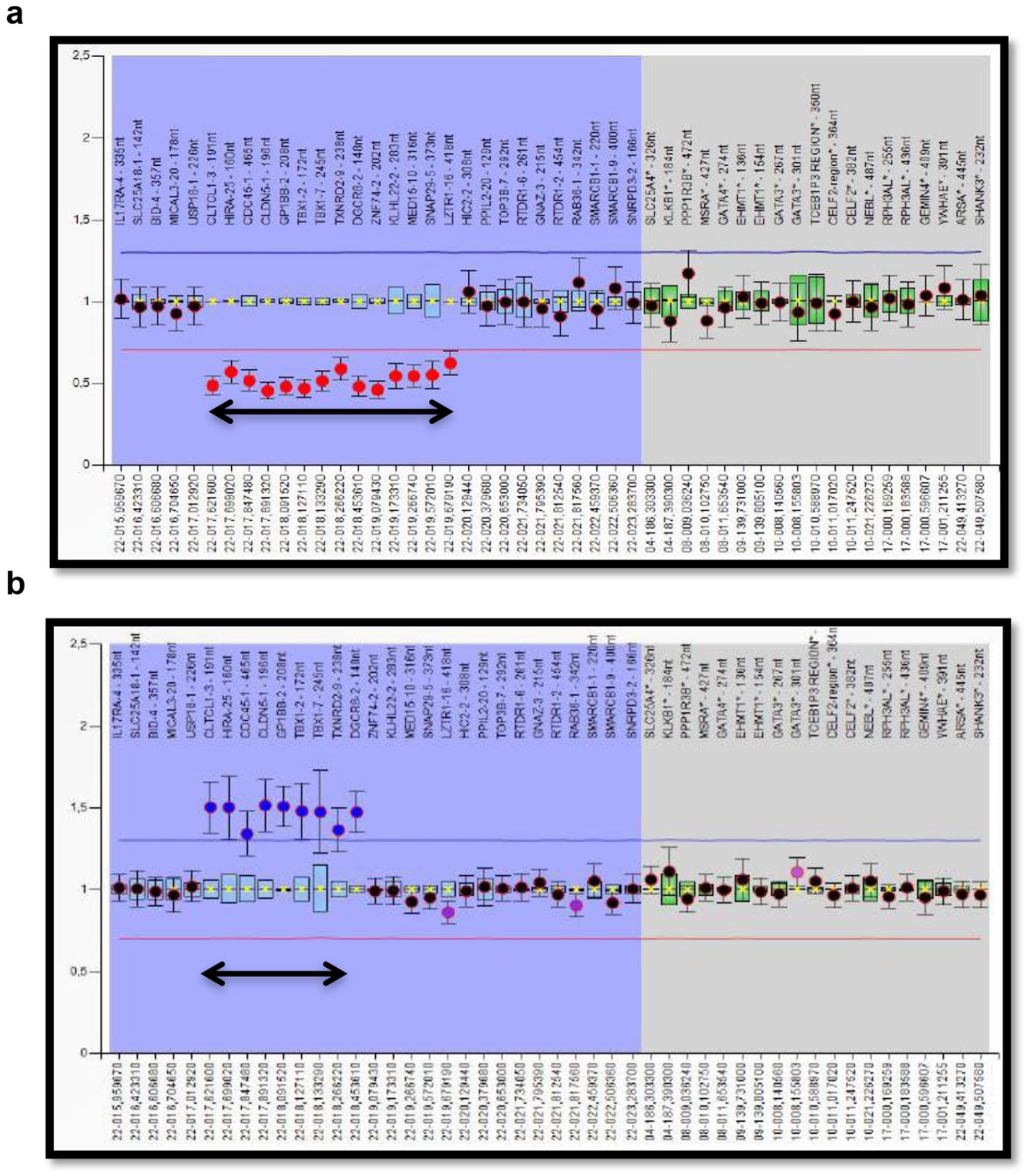
Results of MLPA analysis. Black dots inside the boxes or between the red and blue horizontal lines indicate normal allele, while red dots under the red line indicate a deletion and blue dots above the blue line indicate duplication. (a) Deletion of the typical 3 Mb region in patient 020. The image has been taken from Coffalyzer. (b) 1.5 Mb duplication in patient 027. The image has been taken from Coffalyzer.
MLPA was performed in the parents of 3 cases to establish if the CNV was a de novo or an inherited rearrangement, all of them with a confirmation of a de novo deletion. In the remaining cases, it was not possible to obtain parental samples (Table 1).
Characteristics of patients with CNVs in 22q11.2.

Abbreviation: TDR, typical deleted region.
Characteristics of patients with the 22q11.2 CNVs
Table 1 shows the phenotypic analysis of the 7 patients with CNVs in the 22q11.2 region. None of the patients presented with typical minor anomalies such as cupped ear or bulbous nose. The CHD found in these patients were Interrupted aortic arch Type B and Type C, Ventricular Septal defect (VSD) with the overriding aorta, tetralogy of Fallot and persistent truncus arteriosus. Because most of the patients were in the neonatal period, the CHD was an isolated finding. However, with the clinical follow-up, 2 to 6 months later, some additional findings were reported.
The frequency of 22q11.2 deletion according to the type of cardiac defect is shown in Figure 3.
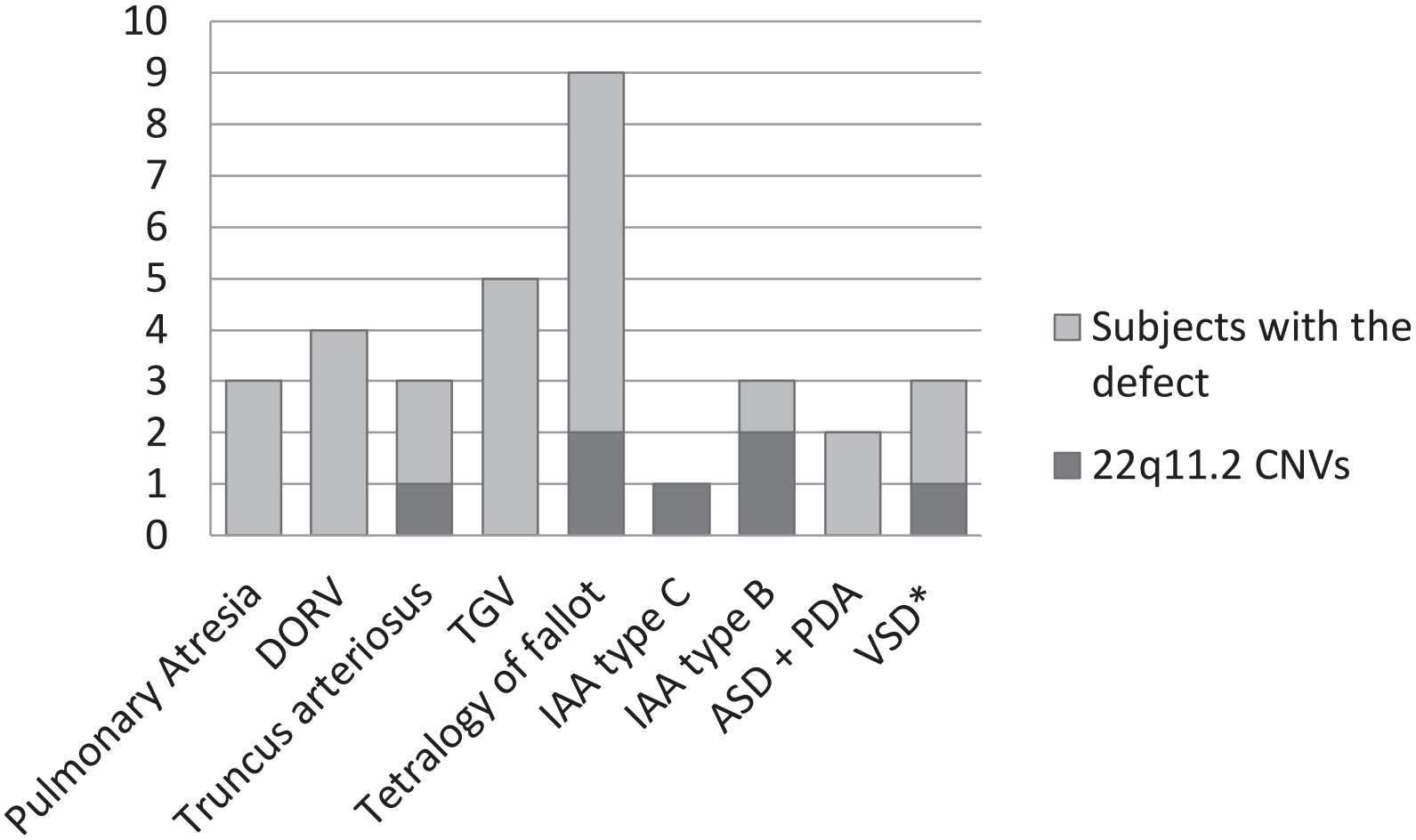
Prevalence of 22q11.2 CNVs according to the type of cardiac disease.
Discussion
Congenital cardiac disease is the most common congenital malformation in humans, 20 it usually presents as an isolated defect and most of the time the etiology remains unknown. 4 Many of the cases have a multifactorial cause, but they can be secondary to chromosomal anomalies, monogenic diseases, microduplications or microdeletions, among others.
CNVs at 22q11.2 are associated with a variety of symptoms including CHD, and thymic, parathyroid, craniofacial developmental and behavioral manifestations. 17 The majority of rearrangements detected in patients in this region are deletions. In this study we found that 21.8% of patients presenting with isolated cardiac defects (at the time of clinical evaluation) have CNVs in the 22q11.2 chromosomal region; most of them had the typical 3 Mb deletion (Table 1).
The 22q11.2 deletion syndrome (also known as DiGeorge or Velocardiofacial syndrome) is the most common deletion syndrome in humans, 20 occurring in 1 to 4 per 6000 live births 21 and is caused by hemizygous micro-deletions on chromosome 22q11.2 22 ; this syndrome has been associated with many different malformations with different severity, commonly includes conotruncal cardiac defects, palatal abnormalities, renal anomalies, immune deficiency (defects in T cells and short telomeres), facial anomalies, developmental delay, learning, and behavioral problems.23-25 The typical combination of hypocalcemia, immunodeficiency, and cardiac defects may lead the clinician to suspect this diagnosis; however, there are cases in which the patient presents with an isolated defect, in which the most reported is the congenital heart disease.7,26 The variation in the clinical findings delays the diagnosis, as evidenced in the study by Cancrini et al 27 in 228 patients with 22q11.2 deletion syndrome. They found that 71% of cases were diagnosed before 2 years of age, mostly due to the presence of CHD and neonatal hypocalcemia. However, the remaining cases in which the patients presented typical facial features, neurodevelopmental and language delay, recurrent infections, and minor heart defects were later diagnosed. These researchers emphasize the importance of an interdisciplinary team for early diagnosis, management, and patient follow-up.
Despite our patients had an isolated CHD at the moment of the clinical evaluation, some of them revealed additional clinical findings after the follow up (2-6 months later); 2 patients were found to be developmental delay and 2 patients reveal thymus aplasia/hypoplasia as a surgical finding. About the developmental delay, it could be considered as a common finding in infants with congenital heart disease before and after surgery, 28 and it could also be a consequence of the 22q11.2 deletion syndrome. Therefore, long-term follow-up of patients is a priority for the timely intervention of clinical symptoms.
Other studies that have tested patients with isolated congenital heart defects for 22q11.2 deletion differ from our data. For example, Fokstuen et al 29 tested 59 patients and Belangero et al 30 tested 6 patients and both found a null frequency for the 22q11.2 deletion. Probably, the differences are due to the molecular method used (FISH instead of MLPA) and the patient age at the moment of the first clinical evaluation (most of our patients were in the neonatal period, and at this moment many syndromic characteristics could not be noticed).
In 2011 in Colombia, Salazar et al 31 published a study of patients with an isolated congenital cardiac defect and they found a prevalence of 4.9% for the 22q11.2 deletion; the lower frequency could be because they included all kind of CHD and because they used a different molecular method (multiplex PCR instead of MLPA). However, our results are very similar comparing them to previous reports of patients with a congenital cardiac defect and additional findings related to the 22q11.2 deletion syndrome.29,32
Among the patients with the deletion in our cohort, the most common cardiac defect was the Interrupted aortic arch, present in the 42,8%. Other anomalies found were tetralogy of Fallot, truncus arteriosus and VSD with the overriding aorta (Figure 1). This data is similar to the previous reports,11,16,29,31-33 highlighting the high incidence of conotruncal anomalies associated with CNVs in 22q11.2. Therefore, prenatal or neonatal diagnosis of conotruncal heart malformation should lead the physician to test the patient for CNVs in 22q11.2.
In the 22q11.2 region, are distinguished 8 LCRs named with letters A through H (LCR22A through LCR22H). Four LCR22 (LCR22A, LCR22B, LCR22C, and LCR22D) are in the 3 Mb region, its hemizygous deletion is found in more than 90% of cases and is associated with the classic phenotypic findings of this syndrome. The second region that is most frequently deleted is a 1.5 Mb situated between LCR22A and B. Finally, the least affected region measures 2.0 Mb and compromises the LCRA trough LCRC. Each region contains different genes, which have been studied to determine a genotype-phenotype relationship that allows a prognosis in affected individuals. 33 These regions have also been reported in duplications. 9 In all cases reported here, the affected 22q11.2 region contains multiple genes and includes TBX11, which in heterozygosis state is associated with cardiac malformations. 34 TBX1 encodes for transcription factor T-box, in this way, the altered regulation of its expression could be related to the variation of the phenotype among patients. 35 On the other hand, studies in mouse models have shown that overexpression of TBX1 causes the same effect on the phenotype as its loss of function18,36; this is consistent with the findings in patients with a duplication in which this gene is included, as in our patient.
Other genetic factors of individuals could also influence the variation in the cardiac phenotype. Mlynarski et al 37 reported the presence of a CNV outside the 22q11 region which contains a duplication of the SLC2A3 gene in patients with CHD and 22q11.2 deletion syndrome; this finding raises the possibility that simultaneous alteration with other CNVs containing important genes in cardiovascular development contributes to the variably of cardiac phenotype.
22q11.2 deletion syndrome usually presents de novo variants. 20 None of our patients had a positive family history of CHD or other major malformation associated with 22q11.2 deletion. We investigated the origin of the variant only in 3 cases, confirming a de novo alteration. (The parental samples could not be obtained in the remaining cases).
The development of different molecular tools has allowed more accurate identification of 22q11.2 CNVs. Molecular techniques most commonly used are fluorescence in situ hybridization (FISH) which identify only the typical deletions, and Multiplex Ligation-dependent Probe Amplification (MLPA) that uses probes capable of hybridizing throughout the 22q11.2 region, allowing the detection of common duplications in this area and both typical (LCR22A-D) and atypical deletions. 38 The Comparative genomic hybridization (array-CGH), is useful when the MLPA is not available or when it is not possible to define a clinical diagnosis.39,40 As previously seen in other reports, we found MLPA to be a useful molecular method that provides an accurate diagnosis of 22q11.2 CNVs. This method let us detect a 1.5 Mb duplication that would be probably missed by other commonly used methods as conventional FISH. 41
The patients from our study were diagnosed at a very early age (0-8 months old) when the congenital cardiac disease was the only single finding in the patient; testing them for the 22q11.2 CNVs lead to an early diagnosis and more specific management and follow up. We also gave the parents a guideline of the 22q11.2 deletion syndrome (Spanish version of the 22q11.2 deletion guideline of the book Preventive Health Care for Children with Genetic Conditions—Cambridge University Press) to aware them about the additional findings that could have their child. 42 This allows the monitoring of the affected children who are followed into adolescence and adulthood and seeks to ameliorated the different neurologic, developmental, psychiatric, and behavioral compromise that they could present, by early medical management.21,43
Conclusions
Our data emphasize the importance of the early testing of 22q11.2 CNVs with MLPA in patients with apparently isolated CHD during the neonatal period, even in the absence of other suggestive symptoms. Early diagnosis positively impacts the management and follow up of the patients.
Footnotes
Acknowledgements
We gratefully thank the patients that participates in the study, the ECLAMC program at San Ignacio Hospital and the different hospitals in Bogotá city which referred the patients.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by Pontificia Universidad Javeriana grants No. ID PPTA 00004318 ID, PRY 004071
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
O.M. and I.Z. conceived, planned the experiments and contributed to the interpretation of the resultd. T.P. carried out the experiments and analysed the data. T.P. wrote the manuscript with support from I.Z., O.M and A.P. J.R. and M.R. contributed to the recruitment and clinical analysis of patients. I.Z. and O.M. supervised de project. O.M. supervised the manuscript.