
Abstract
The cerebellum integrates sensory information and motor actions. Increasing experimental evidence has revealed that these functions as well as the cerebellar cytoarchitecture are highly conserved in zebrafish compared with mammals. However, the potential of zebrafish for modelling human cerebellar diseases remains to be addressed. Spinocerebellar ataxias (SCAs) represent a group of genetically inherited cerebellar diseases leading to motor discoordination that is most often caused by affected cerebellar Purkinje cells (PCs). Towards modelling SCAs in zebrafish we identified a small-sized PC-specific regulatory element that was used to develop coexpression vectors with tunable expression strength. These vectors allow for in vivo imaging of SCA-affected PCs by high-resolution fluorescence imaging. Next, zebrafish with SCA type 13 (SCA13) transgene expression were established, revealing that SCA13-induced cell-autonomous PC degeneration results in eye movement deficits. Thus, SCA13 zebrafish mimic the neuropathology of an SCA-affected brain as well as the involved loss of motor control and hence provide a powerful approach to unravel SCA13-induced cell biological pathogenic and cytotoxic mechanisms.
Zebrafish Cerebellum as a Genetic Model for Spinocerebellar ataxia
The cerebellum is an essential brain compartment that controls motor coordination and learning among other functions to allow us to move smoothly and skilfully. In addition, mounting evidence involves the cerebellum in higher functions such as cognition and processing of emotions. 1 The cerebellum is found in all jawed vertebrates, gnathostomes, 2 and is highly conserved in its cytoarchitecture, connectivity, and function as well as the underlying cellular and genetic mechanisms of differentiation and homeostasis. In zebrafish, the cerebellum develops, differentiates, and reaches functional maturity during embryonic and larval stages during which zebrafish are almost transparent, making them a powerful in vivo system for bioimaging. Moreover, as zebrafish are genetically tractable, they allow for combining molecular genetic studies with in vivo imaging of cell biological and physiological processes making use of the numerous available fluorescent proteins and biosensors.3,4 As zebrafish are becoming an increasingly popular model in the biomedical field, the stage is set to expand cerebellar research in zebrafish to causal investigations of human cerebellar diseases. Spinocerebellar ataxias (SCAs) represent a group of inherited neurodegenerative diseases of the cerebellum that commonly lead to motor discoordination with difficulties in walking smoothly termed ataxic gait. The SCAs belong to a genetically heterologous group caused by more than 40 known distinct genes used for their classification. 5 The SCAs are inherited in an autosomal dominant manner, which prompted us to establish a transgenic zebrafish model affected by a gain of toxicity of a pathogenic SCA allele. As cell type, we chose Purkinje cells (PCs) as these are most commonly affected by SCAs and represent the central neuronal population and sole output of the cerebellum. The PCs integrate external sensory stimuli and motor actions, which are processed into output signals relayed onto efferent neurons usually of the motor system, explaining the difficulties in motor control upon PC degeneration.
PCs in zebrafish develop fast and emerge already at 56 hours postfertilization (hpf) with increasing cells numbers until 7 days postfertilization (dpf). 6 By this stage, PC production reaches a plateau with a population size of about 420 PCs, 6 which is far less than the over 200 000 PCs estimated to populate the mouse cerebellum. At 7 dpf, the cerebellar circuitry in zebrafish is established and PCs like in mammals receive synaptic input from parallel fibres – the granule cell axons – and climbing fibres – axonal connections from the inferior olive. The PC axons form collaterals to other PCs and project either directly out of the cerebellum to terminate on neurons of the octaval nuclei corresponding to vestibular neurons, or they connect to nearby eurydendroid cells, which represent the equivalents to deep cerebellar nuclei neurons in mammals3,7 (Figure 1). Also, the electrophysiological properties of PCs similar to their mammalian counterparts are established at 7 dpf, allowing them to control visual stimuli driven eye or swimming movements like the optokinetic response (OKR) or the optomotor reflex (OMR)3,4 and being involved in motor learning. Optogenetic silencing of PCs consequently impairs OKR and OMR 3 reminiscent of loss of motor control in SCA patients. These findings support the view that zebrafish PCs are well suited to model cerebellar neurodegenerative diseases, as they will allow for continuously monitoring and thus for interconnecting pathogenic molecular processes with anatomical, physiological, and eventual behavioural deficits that mimic clinical signs of SCAs.
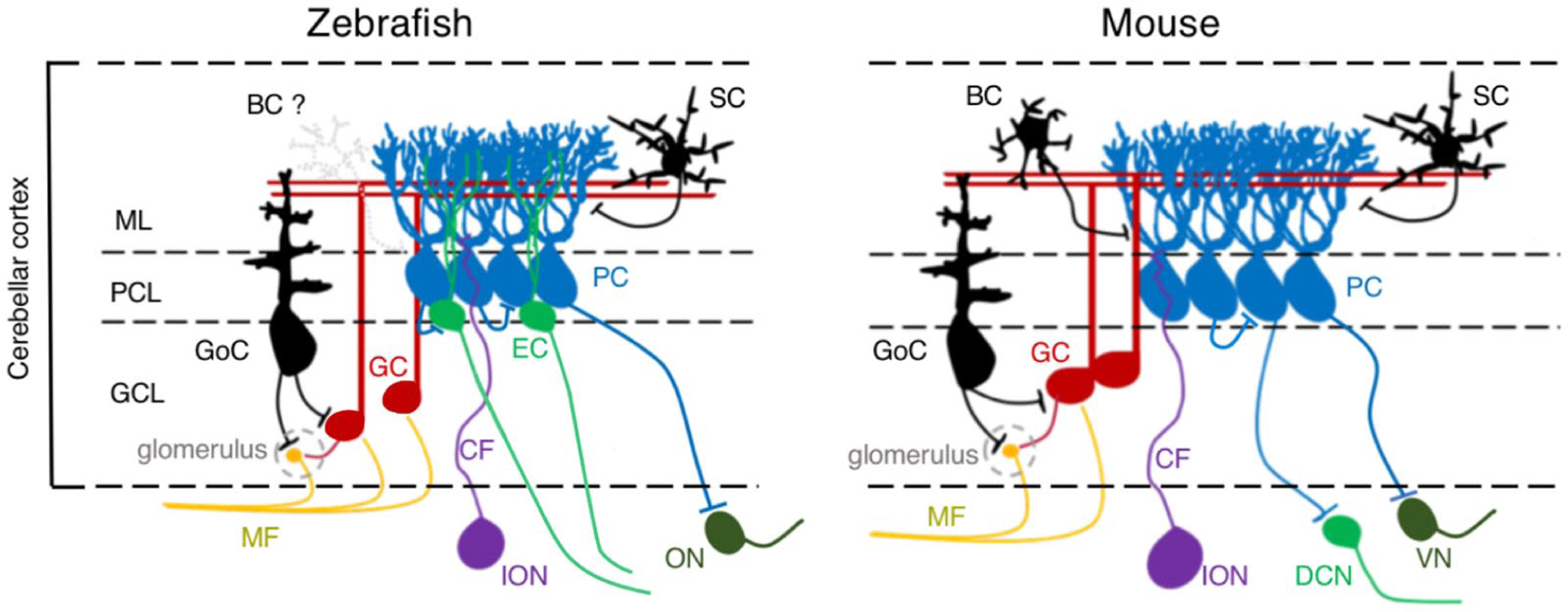
Schematics of cerebellar neurons and their circuitry in zebrafish (left) and mouse (right). BC indicates basket cell; EC, eurydendroid cell; GC, granule cell; GCL, granule cell layer; GoC, Golgi cell; ION, inferior olive nuclei; MF, mossy fibre; ML, molecular layer; ON, octaval nuclei; PC, Purkinje cell; PCL, Purkinje cell layer; SC, stellate cells; VN, vestibular nuclei.
Establishment of a PC-Specific Coexpression System in Zebrafish
For this approach, a prerequisite is the possibility to genetically target PCs in a cell-type-specific manner. We therefore first invested into characterizing a regulatory element upstream of zebrafish carbonic anhydrase VIII (ca8), which is predominantly expressed in differentiating and mature PCs in zebrafish. 6 By transient and stable transgenesis, we first revealed that a 7.54-kb region upstream of the ca8 transcriptional start site was able to drive enhanced green fluorescent protein (EGFP) expression in zebrafish PCs besides expression in a few other tissues such as in the notochord. Subsequently, we narrowed it down to a PC exclusive regulatory element of a size of 258 bp, which we named ca8-derived Purkinje-cell-specific enhancer element (cpce) (Figure 2A to C). Intriguingly, this regulatory element from zebrafish also mediates PC-specific expression in organotypic slice cultures of the mouse cerebellum (Figure 2D), suggesting that regulatory mechanisms of PC-specific expression are conserved among teleosts and mammals. The small size of the cpce further allowed for tandem orientation of several copies of this element, resulting in PC-specific bidirectional expression vectors of different expression strengths when 2, 4, or 8 copies of the cpce were used. The SCA-causing pathogenic alleles can therefore be coexpressed with fluorescent protein reporters at varying concentrations to allow for investigating dose-dependent effects and monitoring disease progression by in vivo time-lapse imaging. Copy numbers of 4 and more cpce elements resulted in increasing ectopic transgene expression in tectal and retinal cells, which could be restricted to PCs again by an miRNA-mediated gene silencing strategy. miRNA181a is predominantly expressed in tectal and retinal cells. Incorporating the miRNA181a target site into the 3′UTR of both transgenes eliminated successfully their expression outside of the cerebellum, while maintaining it at high levels in PCs. This strategy can be easily adopted for other regulatory elements with unwanted expression profiles. Finally, we established a series of PC-specific expression vectors containing 1, 2, and 4 cpce elements, respectively, in bidirectional orientation with different multiple cloning sites and miRNA181a target sites inserted into the corresponding 3′UTR sequences followed by polyA sequences where necessary (4× cpce) (Figure 2E). This vector set will enable fast, dose-dependent, and comparable PC-specific disease modelling in zebrafish accessible for disease monitoring by fluorescent in vivo imaging for SCAs, but also for other diseases affecting PCs such as various types of dystonia, ataxia telangiectasia, Niemann-Pick disease, and autism among others, when making use of PC-specific expression of recombinases, transcriptional activators, and repressors for example.

Zebrafish disease modelling of SCA13 using a Purkinje-cell-specific coexpression system. CCe indicates corpus cerebelli; Val, lateral division of the valvula cerebelli; EGFP, enhanced green fluorescent protein; PC, Purkinje cell.
Neurodegenerative Disease Modelling for Spinocerebellar Ataxia Type 13 (SCA13) in Zebrafish
Using these vectors, we set out to establish a genetic model of SCA13 in zebrafish. This neurological disease is inherited in an autosomal dominant manner leading to cerebellar atrophy. 8 SCA13 is caused by mutations in the KCNC3 allele encoding the voltage-gated potassium channel KCNC3/Kv3.3. Its rodent homologue is expressed in neurons with high-frequency firing rate with prominent expression in cerebellar PCs. 9 It is therefore likely that cerebellar atrophy in SCA13 patients is caused by degenerating PCs as primarily affected neuronal cell type, yet there is currently no in vivo model of SCA13 established, which shows clear signs of neuronal degeneration followed by loss of motor control. Hence, causal analysis of SCA13 is hampered. We initiated SCA13 modelling in zebrafish by analysing the spatiotemporal expression of the zebrafish KCNC3 homologues kcnc3a and kcnc3b. Although kcnc3a was strongly expressed in larval PCs, kcnc3b expression was barely detected in this cell type. Human, rodent, and zebrafish kcnc3 alleles generate a number of splice isoforms terminating in different C-terminal domains of the potassium channel. Splice-isoform-specific mRNA in situ hybridization as well as fluorescence-assisted cell sorting (FACS)-mediated single PC reverse transcription polymerase chain reaction (RT-PCR) revealed that kcnc3a-X12 is expressed at highest abundance in zebrafish PCs. This splice isoform contains the shortest C-terminus of all kcnc3a splice isoforms. We therefore generated a kcnc3a-X12 R335H zebrafish mutant allele (named hereafter kcnc3a R335H ) (Figure 2F) mimicking a mid-age onset SCA13 R420H variant causing progressive cerebellar atrophy in humans. 10
By introducing either zebrafish wild type kcnc3a (kcnc3aWT) or kcnc3a R335H into PC-specific expression vectors coexpressing two fluorescent reporters, membrane-targeted TagRFP-T and nuclear-localized EGFP (Figure 2G), transgenic zebrafish were generated by microinjection into one-cell stage embryos. This allowed for monitoring transgene expressing PCs in the differentiating cerebellum using in vivo confocal microscopy. kcnc3aWT-expressing PCs differentiated indistinguishably from wild-type PCs, increased in number, and developed an elaborated dendritic tree at 7 dpf. In contrast, kcnc3a R335H -expressing PCs displayed progressively increasing signs of axonal and dendritic fragmentation and diminished in cell numbers indicative of progressive neuronal degeneration, which was confirmed by PC counts making use of nuclear fluorescent protein expression (Figure 2H to J). Moreover, when analysed for PC-mediated behaviour, larvae with PC-specific kcnc3aWT expression displayed a normal performance of the OKR, a reflexive eye movement driven by visual stimuli. Consistent with progressive PC degeneration larvae with kcnc3a R335H expressing PCs instead were impaired in proper OKR performance, which is reminiscent of oculomotor disturbance, a clinical hallmark of SCA patients. Taken together, our results strongly suggest that KCNC3 R420H in analogy to its zebrafish homologue kcnc3a R335H induces PC degeneration in a cell-autonomous manner, which appears to be the main cause for the atrophy of the cerebellum leading to ataxic behaviour observed in patients carrying this mutated pathogenic allele.
Molecular Mechanism Underlying SCA13 Pathogenic Mutants induced PC Degeneration
Although the main neuropathological features such as cerebellar atrophy and uncoordinated motor behaviour are shared among SCA13 patients, the clinical symptoms of SCA13 are somewhat heterogeneous as well as the age of disease onset and the course of disease progression. This is explained by allelic heterogeneity as currently more than 10 KCNC3-mutated alleles have been reported so far, ranging from different missense mutations 8 to a C-terminal deletion in the encoded potassium channel. This suggests that the cause of SCA13 is also somewhat heterogeneous influencing the strength of cytotoxic signals and mechanisms in PCs. Electrophysiological studies support this hypothesis, as they indicate that different SCA13 alleles could affect the excitability of neurons to a different extent linked to the disease phenotype. 8
For example, the adult onset KCNC3 R420H mutant allele encodes a non-functional channel subunit mimicked by its zebrafish counterpart kcnc3a R335H . 11 The heterotetrameric wild-type/R420H mutant channels do not result in altering the kinetic behaviour as Kv3.3 channels. This mutant subunit as well as another SCA13 variant R423H, however, mediate a dominant negative effect likely by being trapped along the secretory pathway and mostly in the Golgi together with wild-type subunits12,13 (see later), thereby decreasing the K + current amplitude of the channel.10,11,14 Activation of voltage-gated potassium channels belonging to the family of KCNC/Kv3 (Kv3.1-Kv3.4) depends on unusually positively shifted membrane depolarization to around –20 mV resulting in large K + currents with fast deactivation kinetics allowing neurons to repolarize quickly to sustain high-frequency firing of action potentials. 9 Thus, reduced K + currents as a consequence of a dominant negative effect of KCNC3 R420H or KCNC3 R423H are expected to result in decreased neuronal excitability. Indeed fast-spiking zebrafish motoneurons expressing KCNC3 R420H displayed reduced amplitudes of outward currents and suppressed the excitability of fast-spiking primary motoneurons, thereby decreasing startle behaviour of zebrafish larvae. 15 These altered electrophysiological properties lead to subtle changes in distal axon branches of primary zebrafish motoneurons, when zebrafish kcnc3a R335H is expressed, but did not result in prominent motoneurons degeneration. 16 This is in contrast to the observed PC degeneration in our established zebrafish SCA13 model targeting PCs. 6 Therefore, cytotoxic effects of mutated kcnc3 alleles are neuronal-cell-type specific.
The KCNC3 R423H mutant allele also behaves as a dominant negative subunit, but different to KCNC3 R420H . It affects gating properties of the channels with slower activation and deactivation, when expressed as wild-type/mutant tetramers. 14 This may explain the more severe consequences of KCNC3 R423H -associated SCA13 symptoms occurring already during infant stages. 14 Expression of the respective homologous allele (mouse kcnc3 R424H ) to KCNC3 R423H from mice in primary mouse PC cultures showed reduced outward current densities and broadened action potentials similar to those observed in zebrafish motoneurons affected by KCNC3 R420H expression. 17 Moreover, expression of this mouse homologous mutant allele to human R423H in cultured PCs induced increased intracellular Ca 2+ -levels, likely triggering dendrite shrinkage, and eventually cell death, because these effects could be partially rescued by the treatment with inhibitors of P/Q-type voltage-gated calcium channels. 17 Together, these findings suggest that PCs unlike motoneurons are vulnerable to the expression of dominant negative isoforms of KCNC3, and decreased excitability caused by loss of function of KCNC3 is likely implicated in PC degeneration. Yet surprisingly, while kcnc3 –/– mutant mice show slight gait abnormalities not visible by eye, and PCs in these mutant mice do not show signs of degeneration and cerebellar atrophy, mutant PCs clearly display a reduced excitability 18 resembling a dominant negative KCNC3 isoform expressing neurons. This argues for alternative cell biological mechanisms mediating cytotoxicity in SCA13-affected PCs either acting alone or together with the altered electrophysiological properties.
Further insight was provided by recent cell biological and biochemical studies showing that KCNC3 R420H and KCNC3 R423H are aberrantly glycosylated. In addition, these channel subunits are inefficiently transported to the plasma membrane and are retained to a large extent in the Golgi apparatus.12,13 At least in cells expressing KCNC3 R423H , the epidermal growth factor receptor (EGFR) – in addition to KCNC3 wild-type subunits – is misrouted. Because overexpression of EGFR in Drosophila eyes could rescue KCNC3 R423H induced eye malformation, 13 retention of crucial signalling molecules in the secretory pathway could represent an additional cell biological mechanism of cytotoxicity in SCA13-affected neurons. It would be of interest if KCNC3 R420H affects intracellular membrane transport of EGFR as well, to reveal if this is an allele-specific or common cell biological defect in SCA13-affected cells. The PC-specific coexpression system established by us will allow for testing the involvement of such mis-trafficked molecules in PC degeneration. It will further allow for discriminating whether mis-trafficking alone or in conjunction with altered neuronal excitability leads to progressive cytotoxicity (Figure 3). In addition, recent evidence involved another SCA13 allele KCNC3 G592R in the regulation of actin dynamics in axonal growth cones. 19 As this research was performed on cultured cells, in vivo confirmation could be obtained easily in zebrafish, followed by comparison to implications of KCNC3 R420H and KCNC3 R423H on actin regulation.

Schematic representation of SCA13-induced PC degeneration. KCNC3 R420H and KCNC3 R423H variants show dominant negative effects on WT KCNC3 by being retained in the Golgi apparatus together with WT channel subunits, thereby reducing PC excitability. A possible PC death-associated effector activated by reduced PC excitability could be exerted via increased intracellular Ca 2+ levels likely caused by broadening action potentials excessively activating P/Q-type voltage-gated Ca 2+ channels (VGCC). 17 In addition, the Golgi-retained KCNC3 R423H mutant was shown to impair EGFR trafficking, 13 inducing loss of EGF trophic support, which may be needed for PC survival. EGFR indicates epidermal growth factor receptors; PC, Purkinje cell.
Clearly, the molecular mechanisms causing the SCA13 disease are far from being understood, but distinct research avenues have been opened that await unravelling. Our genetic tools and models for deciphering cell biological and physiological causes of PC degeneration in a cell-autonomous manner directly in vivo making use of coexpression of fluorescent reporters and biosensors will eventually enable clarification of the pathogenic mechanisms underlying PC degeneration in SCA13. Moreover, modelling SCA13 in zebrafish with the possibility to monitor behaviour from single cells to the entire animal offers powerful approaches for in vivo screening and validation of compounds towards counteracting pathogenic molecular and physiological disease mechanisms. Thus, this little fish could make a big splash.
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by funding from Deutsche Forschungsgemeinschaft Grants (KO1949/4-1 and 1949/7-1) to R.W.K.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
KN and RWK wrote this commentary. KN and AD prepared figures. All authors edited the manuscript.