
Abstract
Alterations in the composition of the gut microbiota may be causally associated with several brain diseases. Indole-3-propionic acid (IPrA) is a tryptophan-derived metabolite, which is produced by intestinal commensal microbes, rapidly enters the circulation, and crosses the blood-brain barrier. IPrA has neuroprotective properties, which have been attributed to its antioxidant and bioenergetic effects. Here, we evaluate an alternative and/or complementary mechanism, linking IPrA to kynurenic acid (KYNA), another neuroprotective tryptophan metabolite. Adult Sprague-Dawley rats received an oral dose of IPrA (200 mg/kg), and both IPrA and KYNA were measured in plasma and frontal cortex 90 minutes, 6 or 24 hours later. IPrA and KYNA levels increased after 90 minutes and 6 hours (brain IPrA: ~56- and ~7-fold; brain KYNA: ~4- and ~3-fold, respectively). In vivo microdialysis, performed in the medial prefrontal cortex and in the striatum, revealed increased KYNA levels (~2.5-fold) following the administration of IPrA (200 mg/kg, p.o), but IPrA failed to affect extracellular KYNA when applied locally. Finally, treatment with 100 or 350 mg IPrA, provided daily to the animals in the chow for a week, resulted in several-fold increases of IPrA and KYNA levels in both plasma and brain. These results suggest that exogenously supplied IPrA may provide a novel strategy to affect the function of KYNA in the mammalian brain.
Introduction
The gut-brain axis is receiving increasing attention of neuroscientists since perturbations in the composition and function of the gut microbiota may be causally associated with several neuropsychiatric and neurological diseases.1-4 Products of tryptophan degradation are often suggested to play a major role in this context.5-9 One of these metabolites, indole-3-propionic acid (IPrA), is produced by intestinal commensal microbes via the so-called indole pathway10-12 and enters the circulation of the host. Notably, because of its lipophilic character, IPrA can readily cross the blood-brain barrier and is normally present in the cerebrospinal fluid of both humans and rodents.11,13-15
IPrA possesses potent antioxidant properties 16 and protects neurons against both β-amyloid-induced oxidative stress 17 and ischemic damage.18,19 In view of its ability to enter the brain from the periphery, these neuroprotective effects suggested that IPrA may be of therapeutic use in Alzheimer’s disease, 20 and IPrA is, in fact, currently under clinical investigation for reducing oxidative stress in patients with Friedreich’s Ataxia (ClinicalTrials.gov identifier NCT01898884). Moreover, recent studies revealed that IPrA can be used as a marker for the onset and development of metabolic disorders including type 2 diabetes and non-alcoholic fatty liver disease,21-25 reduces the permeability of the intestinal barrier by pregnane X receptor activation,26,27 and attenuates septic injury and atopic dermatitis by aryl hydrocarbon receptor (AHR) activation.11,28-30
Kynurenic acid (KYNA), an endogenous metabolite of the kynurenine pathway (KP) of tryptophan degradation, has biological qualities which are remarkably similar to those of IPrA.31,32 From a neurobiological perspective, KYNA is known mainly for its neuroprotective, anti-ischemic and anticonvulsant properties,33-36 and for its ability to bi-directionally influence cognitive processes.37-41 These effects have been attributed primarily to KYNA’s inhibition of excitatory amino acid receptors and to its actions as a free radical scavenger.42-44 Notably, however, KYNA also inhibits α7 nicotinic acetylcholine receptor function, 45 can serve as an agonist of the G-protein-coupled receptor GPR35 46 and of the AHR, 47 and acts as a substrate for the organic anion transporters OAT1 and OAT3. 48 KYNA is formed from its immediate bioprecursor kynurenine either through irreversible transamination by kynurenine aminotransferases (KAT I-IV) 49 or by non-enzymatic oxidation. 50 In the healthy mammalian brain, KAT II, which is almost exclusively localized in astrocytes, 51 is the principal enzyme responsible for the neosynthesis of rapidly mobilizable KYNA. 52
In spite of the functional similarities of IPrA and KYNA and their putative biochemical relationship, the effect of IPrA on KYNA formation has not been explored so far. The present study was designed to examine this link in adult rats by either administering a single oral dose of IPrA or feeding animals IPrA for 7 days and then measuring treatment effects on both IPrA and KYNA levels in plasma and brain. In addition, we performed in vivo microdialysis experiments in the medial prefrontal cortex (mPFC) and in the striatum of freely moving rats to study the effects of both systemic administration and local IPrA perfusion on extracellular KYNA concentrations in the brain.
Materials and Methods
Animals
Adult male Sprague-Dawley rats (300-350 g; Charles River Laboratories, Kingston, NY, USA) were used in all experiments. Two animals per cage were housed in a temperature-controlled animal facility (25°C; 40%-60% humidity) on a 12/12 hours-light/dark cycle (lights on at 06:00) with unlimited access to food and water.
Chemicals
IPrA and KYNA were purchased from Sigma-Aldrich (St. Louis, MO, USA). 3H-kynurenine was obtained from Amersham Corp. (Arlington Heights, IL, USA). All other chemicals were acquired from various suppliers and were of the highest commercially available purity.
In vivo studies
Systemic administration of IPrA
Rats received IPrA (50 or 200 mg/kg; dissolved in 0.9% saline) or an equal volume of 0.9% saline by oral gavage (p.o.). Ninety minutes, 6 or 24 hours later, the animals were euthanized in a CO2 chamber and decapitated. Plasma samples and brain tissue (PFC) were rapidly collected from all animals, immediately frozen on dry ice and stored at −80°C until analysis.
In vivo microdialysis
Rats were anesthetized in a chamber filled with 5% isoflurane using a vaporizer and were then mounted in a stereotaxic frame (David Kopf, Tujunga, CA, USA). Anesthesia was maintained during the entire surgery period using a nose mask which continuously delivered 2.0% to 3.0% isoflurane mixed with oxygen. A guide cannula (MAB 2.14.G, SciPro Inc., Sanborn, NY, USA) was then positioned over the mPFC (AP: 3.2 mm anterior to bregma, L: ±0.8 mm from the midline, V: 2.0 mm below the dura) or the striatum (AP: 0.8 mm anterior to bregma, L: ±2.7 mm from the midline, V: 3.5 mm below the dura) and secured to the skull with anchor screws and acrylic dental cement. After surgery, the animals were allowed to recover and were housed individually in acrylic cages with full access to food and water.
On the next day, a microdialysis probe (MAB 9.14.2, membrane length: 2 mm; SciPro) was inserted through the guide cannula. The probe was then connected to a microinfusion pump, and the freely moving rats were perfused with Ringer solution (144 mM, NaCl; 4.8 mM, KCl; 1.7 mM, CaCl2; 1.2 mM, MgSO4; pH 6.7) at a speed of 1.1 μl/min. Microdialysates were collected every 30 minutes. After the collection of baseline samples (1.5-2.5 hours), animals received IPrA or vehicle orally, and sample collection continued for up to 8 hours. Dialysates were divided into two 15 µl samples for IPrA and KYNA measurements, respectively (see below). In separate rats, IPrA, dissolved in Ringer solution (pH 6.8), was applied to the mPFC by reverse dialysis. Microdialysates were again collected every 30 minutes. After obtaining baseline samples (2.5 hours), increasing concentrations of IPrA (10, 100, and 300 μM) were infused in succession (2 hours each). Subsequently, animals received a single dose of IPrA (50 mg/kg) by oral gavage, and sample collection continued for another 2 hours.
Subchronic administration of IPrA
Singly housed rats were fed chow containing IPrA or regular chow (controls) for 7 days. To this end, the compound was mixed into 20 g of chow to allow ingestion of 100 or 350 mg IPrA, respectively, per day. Chow was changed every 24 hours, and the animals had unlimited access to water. On Day 7 (~1 pm), rats were euthanized in a CO2 chamber and decapitated. Plasma and brain tissue (PFC) were rapidly collected and stored as described above.
Analytical procedures
IPrA determination in plasma and brain
Plasma samples were diluted (1:10, v/v) in ultrapure water, and 100 µl of the sample were deproteinized by the addition of 50 µl of 6% perchloric acid. After thorough mixing, samples were centrifuged (16 000×g, 15 minutes). Twenty microliters of the supernatant were applied to a 3 μm C18 reverse phase column (BDS Hypersil; 100 mm × 4.6 mm; Thermo Fisher Scientific, Waltham, MA, USA), and IPrA was isocratically eluted using a mobile phase containing 10 mM sodium acetate, 25 µM EDTA, 0.01% triethylamine and 20% acetonitrile (pH 3.9) at a flow rate of 0.5 ml/min. In the eluate, IPrA was quantified by high-performance liquid chromatography (HPLC) with fluorimetric detection (excitation wavelength: 287 nm; emission wavelength: 340 nm, 2475 fluorescence detector; Waters, Milford, MA, USA). The retention time of IPrA was ~11 minutes.
Brain tissue was homogenized (1:5, w/v) by sonication (Branson Ultrasonics, Danbury, CT, USA) in ultrapure water. Fifty microliters of 6% perchloric acid were then added to 100 μl of the homogenate, and the suspension was centrifuged (16 000×g, 15 minutes). IPrA was determined by HPLC in 30 µl of the resulting supernatant as described above.
KYNA determination in brain
Twenty microliters of the supernatant used for the tissue determination of IPrA (see above) were applied to a 3-µm ReproSil C18 column (100 mm × 4 mm; Dr. Maisch GmbH, Ammerbuch, Germany) to quantify KYNA by HPLC with fluorimetric detection. 53 The retention time of KYNA was ~18 minutes.
Kynurenine and KYNA determination in plasma
Plasma samples were diluted (1:2, v/v, for kynurenine and 1:10, v/v, for KYNA) in ultrapure water, and 100 µl of the sample were deproteinized by the addition of 25 μl of 6% perchloric acid. After thorough mixing, samples were centrifuged (16 000×g, 15 minutes). Twenty microliters of the resulting supernatant were processed by HPLC as described above, and kynurenine and KYNA were quantified by fluorimetric detection. 53 The retention times of kynurenine and KYNA were ~6 and ~18 minutes, respectively.
IPrA and KYNA determination in microdialysate
For IPrA analysis, 15 μl of the microdialysate were applied to a 3 μm C18 reverse phase column (BDS Hypersil; 100 mm × 4.6 mm; Thermo Fisher Scientific), and IPrA was determined in the eluate by the same method used for measurement of the compound in plasma and brain (see above).
For KYNA determination, 15 μl of the dialysate were applied to a 3 mm C18 reverse phase column (BDS Hypersil; 100 mm × 4.6 mm), and HPLC was performed using a mobile phase containing 250 mM zinc acetate, 50 mM sodium acetate, and 4.5% acetonitrile (pH 6.2) at a flow rate of 1 ml/min as previously described. 54 In the eluate, KYNA was detected fluorimetrically (excitation wavelength: 344 nm; emission wavelength: 398 nm; S200 fluorescence detector; Perkin-Elmer, Waltham, MA, USA). The retention time of KYNA was ~6 minutes.
Data were not corrected for recovery from the microdialysis probe.
In vitro studies
KAT II activity
Rats were euthanized, and PFC tissue was rapidly dissected out, frozen on dry ice, and stored at −80°C. On the day of the assay, the tissue was thawed and homogenized in ultrapure water (1:5, w/v; Branson Ultrasonics). After further dilution (1:1, v/v) in 5 mM Tris-acetate buffer (pH 8.0) containing 10 mM 2-mercaptoethanol and 50 μM pyridoxal-5-phosphate, 80 μl of the solution were incubated for 2 h at 37°C in a reaction mixture containing 150 mM Tris-acetate buffer (pH 7.4), 2 μM kynurenine, 0.79 μM 3H-kynurenine (23 nCi), 1 mM pyruvate, and 80 μM pyridoxal-5-phosphate (total volume: 200 μl). Where indicated, IPrA (final concentrations: 10, 30, 100, 300, or 1000 μM) was added to the incubation mixture in 20 μl aliquots (pH 7.0). Blanks contained aminooxyacetic acid (AOAA; 1 mM) in the incubation solution. The reaction was terminated by the addition of 20 μl of 50% trichloroacetic acid and 1 ml of 0.1 M HCl, and the precipitated proteins were removed by centrifugation (16 000×g, 10 minutes). Newly produced 3H-KYNA was then purified by cation exchange chromatography (Dowex 50W; H+-form) and quantitated by liquid scintillation spectrometry. 52
Recombinant human KAT II
Recombinant human KAT II (hKAT II) was prepared as described 55 and stored at −80°C. On the day of the assay, the protein was thawed and diluted 400 times. Five microliters of the solution were then incubated at 37°C for 2 hours in 150 mM Tris-acetate buffer (pH 7.4) containing kynurenine (10 μM), pyruvate (1 mM), and pyridoxal-5′-phosphate (80 μM), in a total volume of 200 μl. IPrA (final concentration: 1, 10, 100, or 1000 μM) was added to the incubation mixture in 20 μl aliquots (pH 7.0). Blanks were obtained by the addition of AOAA (1 mM). The reaction was terminated by the addition of 20 μl of 50% trichloroacetic acid and 1 ml of 0.1 M HCl, and the precipitated protein was removed by centrifugation (16 000×g, 10 minutes). Twenty microliters of the supernatant were applied to a 3 μm C18 reverse phase column (BDS Hypersil; 100 mm × 4.6 mm), and KYNA was determined in the eluate by the method used for the measurement of KYNA in microdialysate (see above).
Data analysis
Data are expressed as the mean ± SEM. Statistical analysis was performed using the Kruskal-Wallis test followed by Dunn’s post hoc multiple comparison test for the effects of time and for subchronic treatment experiments. Two-way ANOVA with Bonferroni’s multiple comparisons test for each timepoint between treatments was used for in vivo microdialysis experiments.
Results
Oral IPrA treatment rapidly and transiently raises both IPrA and KYNA levels in plasma and brain
Endogenous concentrations of IPrA were 6.4 ± 1.7 pmoles/µl in plasma and 0.6 ± 0.2 pmoles/mg tissue in brain. Based on preliminary data (not shown), a dose of 200 mg/kg was selected to assess the effects of IPrA on KYNA in vivo. Oral administration of IPrA raised the levels of the compound in the plasma approximately 51- and 32-fold and in the brain approximately 56- and 7-fold after 90 minutes and 6 hours, respectively. In both plasma and brain, IPrA levels were no longer significantly elevated after 24 hours (P > .05 each; Figure 1a and b).
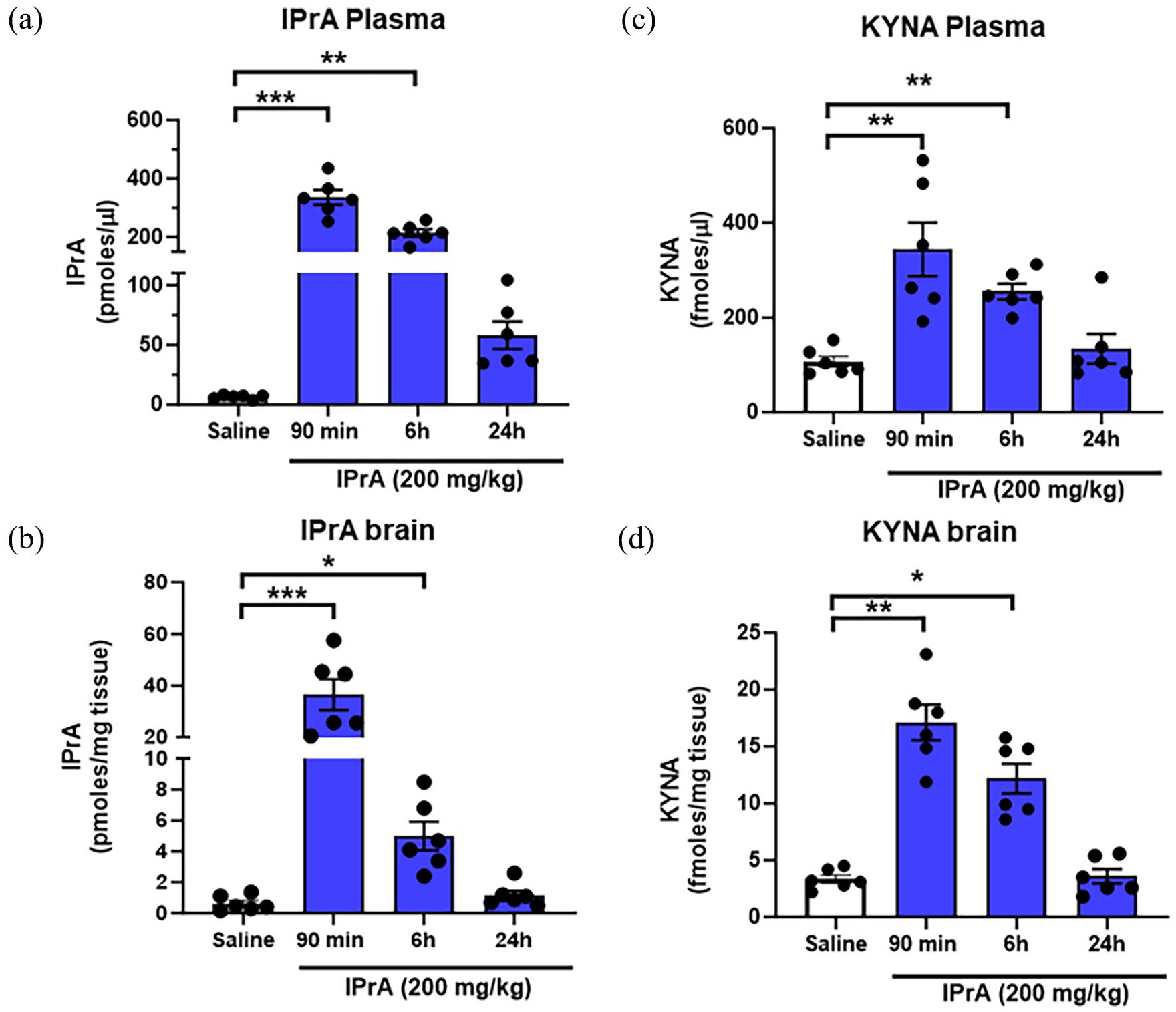
Time-dependent effects of a single administration of IPrA (200 mg/kg, p.o.) on IPrA and KYNA levels in plasma (a, c) and brain (PFC) (b, d). Controls received p.o. vehicle (0.9% saline; only examined after 90 minutes). Data are the mean ± SEM (n = 6 per group; individual data are shown as dots). ***P < .001, **P < .01, *P < .05 versus saline (Kruskal-Wallis test followed by Dunn’s post hoc multiple comparison test).
Measured in the same samples as IPrA, endogenous levels of KYNA were 107.0 ± 11.3 fmoles/µl in plasma and 3.4 ± 0.4 fmoles/mg tissue in the brain. Oral IPrA administration raised the levels of KYNA in the plasma after 90 minutes and 6 hours approximately 3- and 2-fold, and in the brain approximately 4- and 3-fold, respectively, compared to basal levels. As in the case of IPrA (see above), plasma and brain levels of KYNA were no longer significantly elevated after 24 hours (P > .05 each) (Figure 1c and d).
Oral IPrA treatment does not alter kynurenine levels in plasma
Compared to basal levels (3.1 ± 0.3 pmoles/µl) and measured in the same samples as IPrA and KYNA (see above), plasma levels of kynurenine were unchanged at 90 minutes, 6 hours, and 24 hours after systemic administration of IPrA (data not shown).
Effect of oral IPrA on extracellular IPrA and KYNA levels in the mPFC
Compared to basal levels (11.9 ± 2.8 fmoles/µl for IPrA and 3.5 ± 0.2 fmoles/µl for KYNA), the extracellular levels of IPrA and KYNA in the mPFC were significantly elevated beginning at 90 minutes after IPrA administration (200 mg/kg, p.o.), and these increases were sustained up to 5 hours (Figure 2).

Effect of IPrA (200 mg/kg, p.o.) on extracellular IPrA and KYNA levels in the mPFC. Basal levels were collected for 1.5 hours before IPrA administration. Data are the mean ± SEM (n = 6). *P < .05 versus basal levels (two-way ANOVA followed by post hoc pairwise multiple comparisons using Bonferroni’s test).
Effect of oral IPrA on extracellular KYNA levels in the striatum
Compared to basal levels (2.3 ± 0.2 fmoles/µl), oral administration of IPrA also rapidly raised extracellular KYNA levels in the striatum. Similar to the results in the mPFC, oral administration of 200 mg/kg IPrA maintained elevated KYNA levels in the striatum for 8 hours (Figure 3). Additionally, the oral administration of a relatively low dose of IPrA (50 mg/kg) significantly increased extracellular KYNA levels in the striatum, remaining elevated for up to 3.5 hours after administration (Figure 3).

Effect of IPrA (50 or 200 mg/kg, p.o.) or vehicle (0.9% saline, p.o.) on extracellular KYNA levels in the striatum. Basal levels were collected for 2 hours before IPrA or vehicle administration (arrows). Data are the mean ± SEM (n = 5 per group). #P < .05 versus vehicle; *P < .05 versus 50 mg/kg p.o. (two-way ANOVA followed by post hoc pairwise multiple comparisons using Bonferroni’s test).
Effect of local perfusion of IPrA on extracellular KYNA levels in the mPFC
Compared to basal levels (3.2 ± 0.4 fmoles/µl), successive local perfusion of IPrA (10, 100, and 300 µM) did not affect extracellular KYNA levels in the mPFC (Figure 4). Subsequent systemic administration of IPrA (50 mg/kg, p.o.) to the same animals promptly began to raise extracellular KYNA (Figure 4).

Effect of consecutive local perfusions with IPrA (10, 100, and 300 µM) on extracellular KYNA levels in the mPFC. IPrA (50 mg/kg) was subsequently administered to the same animals by oral gavage (arrow). See text for experimental details. Data are the mean ± SEM (n = 4); *P < .05 versus basal levels (two-way ANOVA followed by post hoc pairwise multiple comparisons using Bonferroni’s test).
Effect of subchronic oral IPrA treatment
Endogenous concentrations of IPrA were 8.3 ± 1.4 pmoles/µl in plasma and 1.4 ± 0.5 pmoles/mg tissue in the PFC. Daily feeding with 100 or 350 mg/day of IPrA for a week raised the levels of the compound approximately 19- and 27-fold in the plasma and approximately 2- and 3-fold in the PFC, respectively, compared to basal levels (Figure 5a and b).
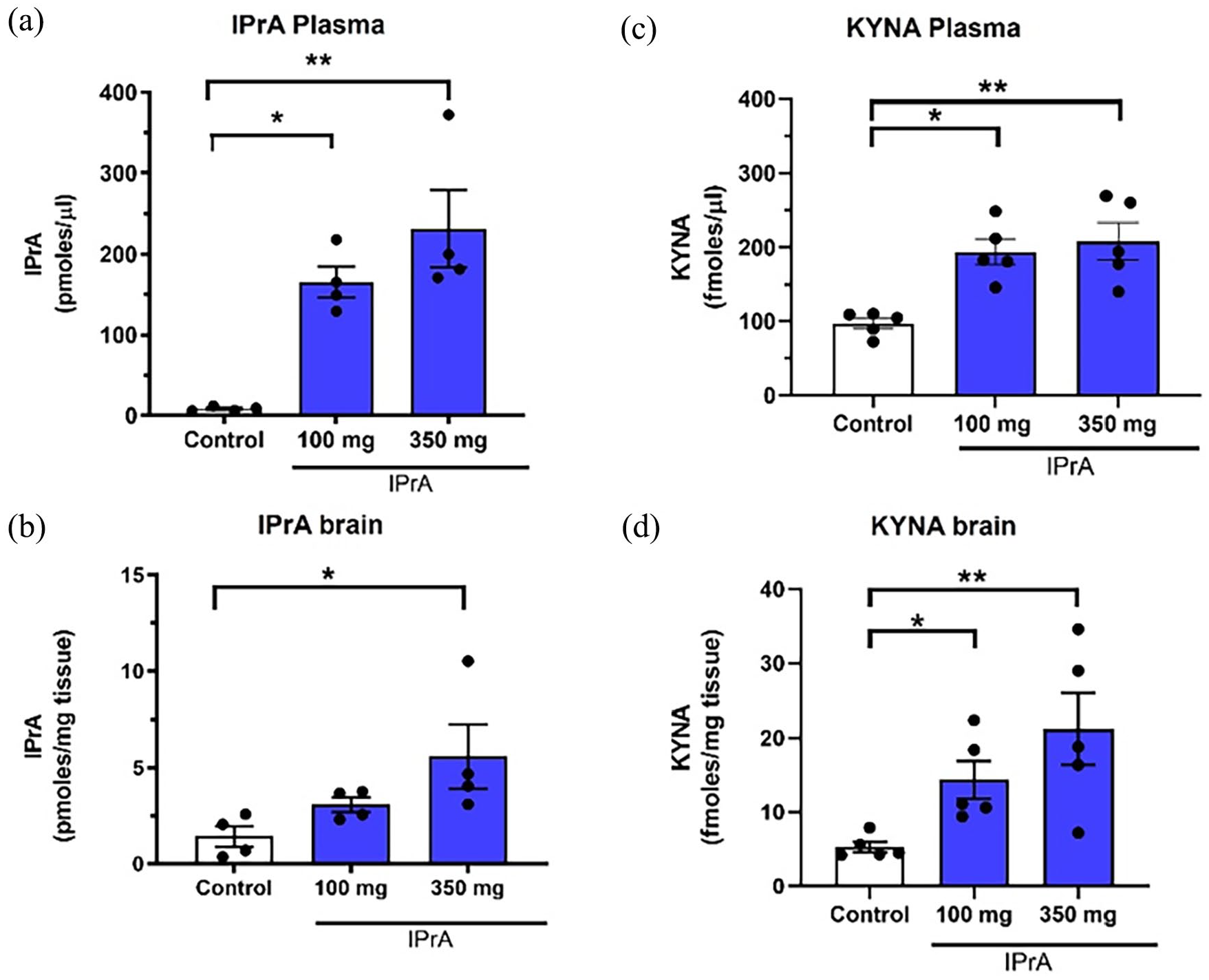
IPrA and KYNA levels in plasma (a, c) and brain (b, d) of rats fed orally with IPrA for 7 days (100 or 350 mg per day). Controls received regular chow. See text for experimental details. Data are the mean ± SEM (n = 4-5 per group). **P < .01, *P < .05 versus the control group (Kruskal-Wallis test followed by Dunn’s post hoc multiple comparison test).
Measured in the same samples as IPrA, endogenous levels of KYNA were 97.3 ± 7.2 fmoles/µl in plasma and 5.3 ± 0.7 fmoles/mg tissue in the PFC. Daily feeding with 100 or 350 mg/day of IPrA for a week raised KYNA levels 2- and 2-fold in the plasma and 2- and 3-fold in the PFC respectively, compared to basal levels (Figure 5c and d).
Effect of IPrA on KAT II activity in vitro
Basal values of KAT II activity in rat PFC tissue homogenate and pure hKAT II were 415.0 ± 41.7 fmoles/h/mg tissue and 0.30 ± 0.07 µmoles/h/mg protein, respectively. In both preparations, addition of IPrA failed to affect KAT II activity up to a concentration of 1 mM (all P > .05; triplicate measures in all cases; data not shown).
Discussion
The present study demonstrated that systemic IPrA administration causes a substantive increase in the brain levels of KYNA, a metabolite well known for its neuroinhibitory, neuroprotective, antioxidative, and free radical scavenging properties.33,42-44 Since IPrA is present endogenously in mammals and can also be detected in the serum of healthy humans (though with high individual variability10,13), this finding may be of physiological significance since even relatively modest elevations of KYNA affect the function of α7 nicotinic acetylcholine and N-methyl-D-aspartate (NMDA) receptors, as well as other receptors including GPR35 46 and the AHR, 47 in the brain. In addition to its role in brain physiology and pathology, which includes regulation of the extracellular levels of classic neurotransmitters (dopamine, glutamate, GABA, and acetylcholine), endogenous KYNA also influences the function of peripheral ion channels and metabotropic receptors,36,56-61 and may thereby play significant roles in metabolic diseases such as diabetes and atherosclerosis.32,62,63 Of special note, physical exercise stimulates KYNA formation in skeletal muscle, 64 which in turn limits insulin resistance and adverse effects linked to inflammatory processes. 65
The biological role of tryptophan-derived indoles has recently gained increasing attention.8,11,66 The enzymatic chain involved in the synthesis and degradation of these compounds in the gut is present in many Gram-negative and Gram-positive bacteria, and there is consensus that IPrA is the terminal product of the catabolic cascade. 67 Thus, gut microbes that express aromatic amino acid aminotransferase catalyze the conversion of tryptophan to indole-3-pyruvic acid, which is subsequently converted to indole-3-lactic acid, further to indole-3-acrylic acid, and finally to IPrA. 68 Notably, IPrA modulates microbiota composition in the gut, inhibiting gut dysbiosis 23 and regulating gastrointestinal barrier function. 26 However, gut-derived IPrA also readily enters the circulation and affects the function of several peripheral organs (see Jiang et al 11 for review) and, as a lipophilic compound, easily crosses the blood-brain barrier. 13
Interest in the fate and function of IPrA is based on the remarkable, and remarkably complex, biological effects of the compound observed in pre-clinical and clinical studies. Thus, IPrA facilitates regeneration and functional recovery of sensory axons through an immune-mediated mechanism,69,70 promotes intestinal homeostasis,71,72 boosts muscle tissue development and reduces muscle cell inflammation, 73 modulates mitochondrial function in cardiomyocytes, 74 mediates abnormal synaptic pruning of hippocampal microglia, and affects susceptibility to autism spectrum disorders. 75 In the brain, IPrA can attenuate neuronal damage and oxidative stress,17,18,76 and modulates inflammation and immunity via AHR activation and its downstream effects.8,77 Of note, elevated IPrA levels are associated with cognitive impairment. 78 Taken together, IPrA impacts an array of immune, gastrointestinal, cardiovascular, and central nervous system functions in mammals. In several cases, these phenomena have been convincingly linked to the antioxidant properties of the metabolite and to its ability to inhibit the synthesis of proinflammatory cytokines (cf. Introduction).
A considerable number of biological effects of IPrA, both within and outside the gut, are qualitatively similar to those associated with elevated KYNA levels. Our present demonstration that either acute or subchronic oral IPrA application increases KYNA levels both in the plasma and in the brain therefore raises the question if and to what extent KYNA may play a substantive role in the beneficial and/or adverse consequences of endogenously produced or—as in the present study—exogenously administered IPrA. In other words, could (some of) the biological properties of IPrA be causally related to its ability to serve as a direct or indirect bioprecursor of KYNA?
Previously published data, as well as results from the present study, do not provide straightforward answers to this question. Both in vivo and in vitro studies in mammals have so far failed to show that IPrA, unlike its related metabolite indole-3-pyruvic acid, can be oxidized non-enzymatically to form KYNA 8 (Poeggeler, unpublished observations). Moreover, we showed here that IPrA, when applied locally in vivo, had no impact on KYNA formation in the brain and does not affect KAT II activity, which accounts for ~75% of KYNA neosynthesis in the adult rat brain, 52 in vitro. In fact, IPrA inhibits 2 KP enzymes upstream from KYNA, namely tryptophan-2,3-dioxygenase 79 and KAT I, 80 and prevents the production of the pivotal KP metabolite kynurenine in human astrocytes. 81 Also of note in this context, some central effects of IPrA, such as its ability to protect against hydrogen oxide-induced oxidative damage in vivo, are seen after either systemic or intracerebral application of the compound. 16 Thus, although we cannot rule out that KAT III and/or KAT IV may play a role,49,51 mechanisms unrelated to KYNA neosynthesis from kynurenine can clearly account for some of IPrA’s effects within the brain. 82 Taken together, functionally significant connections between IPrA and KYNA ought to be explored further and may reveal, for example, the IPrA-induced peripheral formation of currently unknown, brain-penetrant compounds, which could secondarily raise KYNA within the brain. Future studies should also be designed to examine the possible effects of systemic IPrA administration on the levels of neuroactive KP metabolites other than KYNA, including 3-hydroxykynurenine, xanthurenic acid, cinnabarinic acid, picolinic acid, and quinolinic acid, 83 and to evaluate IPrA-induced KP changes in additional brain regions.
Pharmacological approaches to up- or down-regulate KYNA function in the brain are increasingly considered as therapeutic options in major neurological and psychiatric diseases.84-87 In view of the fact that IPrA can be safely administered to humans even at high doses, 88 the translationally most relevant question generated by the present study is therefore whether systemically produced or exogenously applied IPrA, irrespective of its precise mechanism of action, predictably mimics—or is additive with—certain characteristics of KYNA function and dysfunction in the brain. In these cases, targeted genetic or pharmacological up- or down-regulation of IPrA levels in microbes or other cells could be expected to provide interesting, conceptually novel strategies for the treatment of a number of major brain disorders.
Footnotes
Acknowledgements
The authors thank Ms. Marian Thomas for outstanding technical assistance.
Author Contributions
Conceptualization, K.V.S., S.E., B.P., and R.S.; methodology, K.V.S., T.B.-A., and L.S.; software, Y.Z. and M.T.-A.; validation, K.V.S., L.S., and S.E.; formal analysis, K.V.S., L.S., and M.T.-A.; investigation, T.B.-A., Y.Z., and M.T.-A.; resources, S.E. and R.S.; data curation, K.V.S., T. B.-A., Y.Z., and M.T.-A.; writing: original draft preparation, K.V.S., T.B.-A., and Y.Z.; writing: review and editing, K.V.S., L.S., S.E., B.P., and R.S.; visualization, K.V.S., T.B.-A., Y.Z., and M.T.-A.; supervision, L.S., S.E., and R.S.; project administration, K.V.S. and T.B.-A.; funding acquisition, S.E. and R.S. All authors have read and agreed to the published version of the manuscript.
Declaration of conflicting interests:
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: R.S. is co-founder of Kynexis BV, which develops kynurenic acid-related compounds for human use.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by NIMH grant P50 MH103222 (Silvio O. Conte Center for Translational Mental Health Research) and The Swedish Research Council (2021-02251).
Institutional Review Board Statement
Experimental protocols were approved by, and performed in accordance with, the guidelines of the Institutional Animal Care and Use Committee of the University of the Maryland School of Medicine (Baltimore). All efforts were made to minimize the number of animals used and to optimize their well-being.
Data Availability Statement
Data are contained within the article.