
Abstract
Protein dynamics governs most of the fundamental processes in the human body. Particularly, the dynamics of loops located near an active site can be involved in the positioning of the substrate and the reaction mechanism. The understanding of the functioning of dynamic loops is therefore a challenge, and often requires the use of a multi-disciplinary approach mixing, for example, crystallographic experiments and molecular dynamics simulations. In the present work, the dynamic behavior of the JK-loop of the human indoleamine 2,3-dioxygenase 1 hemoprotein, a target for immunotherapy, is investigated. To overcome the lack of knowledge on this dynamism, the study reported here is based on 3 crystal structures presenting different conformations of the loop, completed with molecular dynamics trajectories and MM-GBSA analyses, in order to trace the reaction pathway of the enzyme. In addition, the crystal structures identify an exo site in the small unit of the enzyme, that is populated redundantly by the substrate or the product of the reaction. The role of this newer reported exo site still needs to be investigated.
Keywords
Introduction
Loop dynamics in proteins structures is essential in the description of enzymatic mechanisms,1-3 and can be studied by crystallography.4,5 By collecting structures with intermediate ligand positions, it has been possible to understand the organization of dynamic loops.1-3 These advances play an important role in the development of effective drug molecules for many therapeutic targets. 6 However, although crucial to a deep understanding of the enzyme mechanism, the dynamic nature of flexible loops is also a well-known challenge to crystal refinement. This is why it is common to use a multi-disciplinary approach, for example, mixing crystallography and molecular dynamics, to study these phenomenons. 7
In this context, the hemoprotein human indoleamine 2,3-dioxygenase 1 (hIDO1) is involved in the first step of the kynurenine pathway, aimed at the degradation of L-Trp into a series of metabolites (L-kynurenine, redox cofactors, neuroprotectors, and neurotoxins). This enzyme is a well-known therapeutic target in immunotherapy.8-10 In particular, overexpression of hIDO1 by cancer cells has been shown to induce immune escape and resistance to immunotherapy11-14 by decreasing T cell activation.11,13,15-18 Although more than 50 crystal structures exist for this therapeutic target,19-37 only partial structures of the active site, that is, without the refinement of the dynamic JK-loop, were obtained so far. The JK-loop (residues 360-380) of hIDO1 is a dynamic loop located at the entrance of the active site, in contact with the heme cofactor. For the last 5 years, it has been shown that it is involved in the closure of the active site and, consequently, in the inhibition of the enzyme. 38 The difficulty in refining the JK-loop comes from the high flexibility of its N-terminal part (residues 360-374). The C-terminal part (residues 375-380) is less flexible and is stabilized, as reported in most of the published hIDO1 structures. Depending on the presence of L-Trp or inhibitors, the JK-loop can adopt 3 interconvertible conformations, that is, opened, closed, and intermediate. Recently, due to a change in the crystal packing and despite its agitation, the complete JK-loop was refined for the first time in the absence of ligand. 39 The structure shows an intermediate form of hIDO1, and the heme cofactor is located inside the enzyme, involving an electrostatic interaction with the residue K377. Since the information on the conformation of the loop is recent, the folding mechanism remains still unclear. To clarify the latter, it would be necessary to obtain the conformation of the JK-loop according to several intermediate positions of L-Trp when it enters the active site. Although several reaction snapshots showing the protein in the presence of L-Trp were recently generated by molecular dynamics, 40 the first intermediate position of L-Trp in crystallography was obtained by Pham et al 30 in 2021. However, the dynamic loop was still only partially refined in their structure. The other lack of structural information on hIDO1 comes from the potential identification of an exo site, whose existence was postulated for the first time by Greco et al 40 in 2019. Although the affinity of L-Trp for this site is similar to the one observed in hTDO, it has never been demonstrated by crystallographic experiments. Thus, the study of L-Trp along the reaction pathway characterized by the fully refined JK-loop and the identification of the exo site are the next steps to improve the understanding of the therapeutic target and help to the design of new drugs.
In the present study, 3 new crystal structures of hIDO1 (resolution values of 2.30, 2.50, and 2.50 Å) were refined in ferrous state with L-Trp inside the active site. L-Trp is observed in different positions allowing to trace its entry in the active site as well as the evolution of the dynamic loop during this process. To provide more information on the folding dynamics of the protein in solution, these structures were simulated using 300 ns Molecular dynamics (MD) calculations. Finally, in order to characterize the influence of the conformation of the dynamic loop on the affinity of heme and/or L-Trp, MM-GBSA calculations were performed.
Experimental Section
Production and purification
Escherichia coli BL21 (DE3) cells were transformed with a pET-28a plasmid provided by Tong and co-workers 27 containing the coding gene for hIDO1 with 2 surface mutations (K116A and K117A). The production and purification of the enzyme was performed according to the protocol already described in the literature. 39 After purification, the protein was exchanged into crystallization buffer consisting of 5 mM HEPES/NaOH pH 7.6, 200 mM NaCl, 5 mM DTT, and concentrated at a concentration of 16 mg/mL using Amicon filters with a 10 kDa molecular-weight cut-off membrane. The hIDO1 protein is stored at −80°C.
Co-crystallization assays
Before proceeding to co-crystallization assays, the reduced protein was incubated on ice with L-Trp at a final concentration of 2 mM for, at least, 15 minutes. After incubation, using the hanging-drop vapor diffusion method at 293 K, drops were made with 1 of the well solution and 1 of the protein/L-Trp solution. The wells contained a solution consisting of 14% to 15% polyethylene glycol 3350, 0.1 M KH2 PO4/NaOH at pH 6.25 to 6.50. The red crystals appeared in 1 day and were left for at least 2 weeks before the diffraction experiment and data collection. Prior data collection, crystals were soaked for at maximum 2 min in well solution supplemented with a cryo-solution at a final concentration of 20% glycerol and 20 mM sodium dithionite. The crystals were subsequently flash-cooled in liquid nitrogen.
X-ray collection and data refinement
The resources of the SOLEIL synchrotron (Gif-sur-Yvette, France) allowed the collection of data on the PROXIMA-2 (PX2) beamline using a Dectris EIGER X 16 M detector at 100 K and at a single wavelength of 0.980 Å. Data indexation was performed with XDS suite. 41 Molecular replacement using Phaser 42 and a monomer from PDB entry 7A62 as the model solved the phase problem. Model building and structure refinement were performed using Coot 43 and Phenix. 44 The heme cofactor, L-Trp, and NFK molecules were added to the monomers using eLBOW. 45 Three crystallographic structures showing different positions of L-Trp in the active site of hIDO1 have been obtained. For visualization purposes, crystallographic maps (2Fo-Fc) are cut around the ligands using adequate Phenix module and a model cut method. 44 All figures were generated using PyMOL (version 0.99; Schrodinger) 46 or Maestro for the interaction maps.
Binding site studies
The enzyme binding sites were determined with the SiteMap tool of the Maestro suite. 46 For this purpose, the crystal structures were prepared with Protein Preparation Wizard tool.46,47 The different ligands were removed manually in order to leave the pockets empty. The analysis was run to determine at least 10 top-ranked binding sites with 10 site points per site using a fine grid method.
Model construction and molecular dynamics studies
To study the dynamics of the JK-loop of hIDO1, the crystal structures 7P0R and 7P0N were completed by means of a homology model. The model was constructed with the Maestro program using Prime and the Structure Prediction Wizard with the sequence from the structure 7A62 as the starting model.46,48,49 The heme cofactor and the L-Trp molecule were then added to the resulting homology model at a position observed by crystallography. The resulting structures were used as starting points for the MD simulations in complement to the structure hIDO1-closed (7NGE). Each monomer with an L-Trp molecule in the active site was used as the starting model. MD simulations of the different protein/ligand complexes were run using GROMACS 2020 50 with the CHARMM27 force field 51 and CMAP corrections for the protein. H atoms were added using GROMACS. The ligand-specific topology files were created using the SwissParam software 52 based on the position in the protein and the protonation state of the molecule at pH 7.0. The L-Trp molecule is allowed to move from its original position during the simulations. The solvatation, the optimization, the equilibration, and the production stage of the models were performed as previously described in the literature. 39
MM-GBSA analysis
In order to evaluate the affinity of L-Trp and the heme cofactor depending on the different crystal structures, MM-GBSA calculations were performed using the Maestro Prime tool.46,48,49 The method was applied to the 3 crystal structures containing the ligand. For the calculation of the affinity of the heme cofactor, the structure without any ligand was also used (PDB code: 7A62). The monomers from the PDB structures were first prepared with the heme group and, if present, the ligand inside by the Protein Preparation Wizard tool of the Biologics suite from Maestro program.46,47 In order to properly define the coordinative bonds between the iron ion and the nitrogen atoms of the porphyrin ring, a preprocessing of the macromolecule is performed by creating zero-order bonds to the metal. The addition of hydrogen atoms is also done at this stage with a validation of the protonation states by the program Epik.46,53,54 The ferrous redox state was retained for iron in agreement with the crystallization conditions. The final model was optimized with the OPLS3e force field.46,55 Separate structure files were then obtained for the ligand and/or the cofactor and the enzyme. The MM-GBSA calculations were performed using the VSGB solvatation model46,56 together with the OPLS3e force field. The enzyme was kept rigid to preserve a conformation close to the crystal conformation. Prior to the calculation using Prime MM-GBSA, validation tests on the system against known data have been done using human hemoglobin and sperm whale myoglobin to validate the ranking of affinity for the heme cofactor (data not shown).
Results and Discussion
Positions of L-Trp inside the active site
After the analysis of a large amount of hIDO1/L-Trp co-crystals in ferrous condition using synchrotron radiation and the refinement of the data, 3 crystal structures, presenting different L-Trp positions in the active site, were obtained (7NGE, 7P0R, and 7P0N). For the clarity of the following manuscript, the structures will be renamed hIDO1-closed, hIDO1-intermediate, and hIDO1-opened, respectively. The structures (statistical data are shown in the Supplemental, Table SI1) present hIDO1 as a dimer of dimer, as already mentioned in the literature. 39 The protein crystallizes in a holo form, with a heme group in each monomer of the asymmetric unit. Molecules of L-Trp are observed around the protein and inside the active site of 3 monomers, 1 for each structure. Since crystallization takes place under reducing conditions, it is also possible to observe the product of the reaction between hIDO1 and L-Trp, N-formylkynurenine (NFK), outside the enzyme. The position of the L-Trp molecules in the 3 active sites relative to the heme cofactor is shown in Figure 1.

General view and zoom on the position of the residues in the active site with respect to L-Trp in structure: (a) hIDO1-closed (
In the structure hIDO1-closed (Figure 1a), L-Trp (occupancies = 68%) is located in the so-called reaction position of the monomer A. As previously mentioned in the literature 24 (Supplemental Figure SI1), the aromatic part of L-Trp occupies the pocket A of the active site. The indole site is stabilized by the hydrophobic core involving Y126, F163, F226, L234, G262, and A264. S263 forms a hydrogen bond with the nitrogen atom of the indole ring. The polar part of L-Trp is positioned in the pocket B of the active site. The residues R231, K373, and K377 bring positive charges and stabilize the carboxylate part of L-Trp. To allow this interaction, the JK-loop is closed, as detailed later. The protonated amine group of the substrate directly interacts with the carboxylate group of the heme cofactor.
In the structure hIDO1-intermediate, the substrate is located in the monomer B (occupancies = 86%), in a position that is close to the one recently described by Pham et al 30 (Supplemental Figure SI2a). However, unlike Pham et al no photochemical intermediate is observed in the present case. The currently observed position is thus a temporary position showing the entry of L-Trp into the active site. It mainly occupies pocket B, opened by the intermediate conformation of the JK-loop. The heme cofactor interacts with the nitrogen atom of the indole group and participates to the stabilization of the polar part of L-Trp by means of a hydrogen bond with a water molecule in interaction with the protonated amine. The carboxylate part of L-Trp is stabilized by the main chain of G236 and K238. In this case, K373 and K377 are not directly involved in the stabilization of the polar part.
The structure hIDO1-opened (Figure 1c) contains L-Trp in monomer C with an occupancies of 87%. The molecule is not positioned in a reactive position but in another intermediate position which is more upstream of the active site. This position has never been reported in any other crystal structure but is very close to the metastable positions resulting from the MD analysis of Greco et al. 40 Consequently, L-Trp lies in pocket B and does not have access to pocket A. The stabilization of the indole ring of L-Trp is mainly achieved by the heme cofactor. Concerning the polar part, the heme cofactor interacts directly with the protonated amine while R231 is involved in an electrostatic interaction with the carboxylate function. In the present case, the JK-loop is located too far from the active site to contribute to an interaction with the substrate or the cofactor. On the whole, the heme conformation is not totally planar in order to interact with the ligand in the case of the 2 temporary intermediate positions. Such a distortion is known for other hemoproteins but has never been highlighted for hIDO1. 57
Conformation of the JK-loop according to the L-Trp position
Depending on the relative position of L-Trp with respect to the active site and the heme cofactor, the dynamic loop adopts different conformations. In the case of the hIDO1-closed structure (Figure 2a), a closed loop is observed with very little surface opening (Figure 2b). The mean B-factor for the loop in a closed position in presence of L-Trp amounts to 94 Å2. This value is 16 Å2 lower than the ligand-free structure (PDB code: 7A62, 110 Å2), and is totally acceptable in comparison to the average of B-factor on the whole structure (56.5 Å2). It suggests that the presence of L-Trp in the reaction position and the closure of the loop lead to a huge stabilization of the loop compared to the previously published ligand-free structure. 39 In such a closed conformation of the C-terminal part of the loop, K377 adopts 2 distinct conformations according to the location of L-Trp (Figure 2c). When L-Trp is inside the active site, K377 is oriented toward the substrate (K377 (A) in green with an occupancy of 83%) which is stabilized. The distances between the nitrogen atom of K377 and the 2 oxygen atoms belonging to the carboxylate group of L-Trp amount to 3.0 and 3.6 Å. In this position, K377 brings a positive charge near the carboxylate of the cofactor. The distances between the nitrogen atoms of K377 and the oxygens from the carboxylate of the heme cofactor amount to 3.1 and 4.8 Å. Otherwise, K377 (K377 (B) in gray with an occupancy of 17%) interacts only with the cofactor at distances between the nitrogen atom and the 2 oxygens of 4.0 and 4.7 Å, as observed in the case of the substrate-free structure. Thus, the loop has a dual role, both stabilizing the substrate and the cofactor.

JK-loop in monomer A of hIDO1-closed structure: (a) relative conformation of the loop with respect to the active site, (b) surface of the protein with the closure of the loop, and (c) alternative conformations of K377 with an interaction between K377(A) (green) and L-Trp or an interaction between K377(B) (gray) and the heme cofactor.
In the temporary intermediate positions (Figure 3a, I), a first possible conformation shows that the loop is no longer closed but is partially open, as already described by Pham et al 30 (Supplemental Figure SI2b). It is explained by the position of the ligand which has not yet fully entered the active site. The loop has not yet fully closed. Consequently, the agitation of the loop increases due to its partial opening. The presence of the ligand in a temporary intermediate position leads to a less stable JK-loop with a mean B-factor of 114 Å2 for the refined C-terminal part. The C-terminal part remains close to the active site, allowing the electrostatic interaction of the protonated amine from K377 with the carboxylate group of the cofactor, at a distance of 5.2 Å (Figure 3a, II). As already mentioned in the case of the structure without ligand, 39 this interaction confirms the role of lysine K377 to maintain the cofactor in its pocket during the positioning of the substrate and to avoid lability of the latter. Therefore, the loop does not have any longer a role in stabilizing the substrate. In this case, due to a larger agitation, the N-terminal part is too disordered to be refined. It may be caused by the rapid positioning of L-Trp, resulting in a fast rearrangement of the loop at the N-terminus. As a consequence, the protein surface is much more open than in the case of hIDO1-closed (Figure 3a, III). In the second possible conformation (Figure 3b, I), the more upstream position of L-Trp leads to a fully opened JK-loop as indicated by the fragment composed of residues T379 to T382 (Figure 3b, II). Since the ligand is not well positioned in the active site, the loop is not well refined, even at the C-terminus. The surface is widely open (Figure 3b, III) and the A-site is hardly accessible to the ligand at this stage of the enzymatic reaction.

Conformation of the JK-loop in different states of the active site. (a) I, Conformation of the JK-loop in hIDO1-intermediate structure according to the ligand position. II, Role of K377 for the interaction with the heme cofactor. III, Van der Waals surface resulting of the partial opening of the JK-loop. (b) I, Conformation of the JK-loop in hIDO1-opened according to the ligand position. II, Positioning of the C-terminal part away from the ligand. The refined fragment is from T379 to G380. III, Van der Waals surface resulting from the total opening of the JK-loop.
Exploration of the behavior of the JK-loop by molecular dynamics
Molecular dynamics (MD) simulations were carried out to study the behavior of the solvated dynamic loop depending on the different ligand positions. The analysis of the 300 ns hIDO1-closed MD trajectory highlights that L-Trp does not significantly move during the simulations (Figure 4a and b). Consequently, the distance between the protonated amine of L-Trp and the carboxylate group of the cofactor amounts to 3.6 ± 1.0 Å. This is close to the distance of 3.0 Å observed experimentally by crystallography. The conformation of the loop observed from a 300 ns-MD trajectory with L-Trp is consistent with the starting closed-structure resulting from crystallographic experiments. Nevertheless, a slight induced fit is observed (Figure 4a) wherein the last turn of the K helix is deconstructed in contrast to the experimental structure. It leads to a complete closure of the protein surface by bringing the JK-loop closer to the helix F (residues 286-300). Residue T379 directly interacts with residue D394 in the helix F (Supplemental Figure SI3). The MD simulation confirms the presence of an interaction, described by Greco et al 40 between the JK-loop and the helix EF. Due to the complete closure, the K377/ heme interaction is lost at 300 ns. However, it is hypothesized that the closure preserves enough heme inside the active site to avoid the need of the stabilizing K377/heme interaction. During the MD simulation, K377 moves to assist the stabilization of the substrate. Particularly, the distance between the 2 oxygen atoms of L-Trp and K377 varies over the 300 ns, with an average of 4.1 ± 1.1 Å for O1 and 5.8 ± 1.2 Å for O2 from L-Trp, in comparison to the 3.0 and 3.6 Å observed in the crystal structure. On the whole, the results of the simulations confirm the stabilization of the loop observed in crystallography. Indeed, the RMSF analysis of the Ca atoms (Figure 4c, green) of the protein shows a stable and non-agitated loop for the C-terminal part compared to the results already published for the protein without ligand (Figure 4c, black). 39 When a MD simulation is performed without L-Trp but with the same starting close conformation for the loop, the agitation of the C-terminal part is recovered (Figure 4c, pink). This agitation increase comes from a reopening of the enzyme, leading to a conformation of the intermediate JK-loop in the absence of ligands (data not shown). It thus suggests that the ligand is essential for a stabilization of the JK-loop.
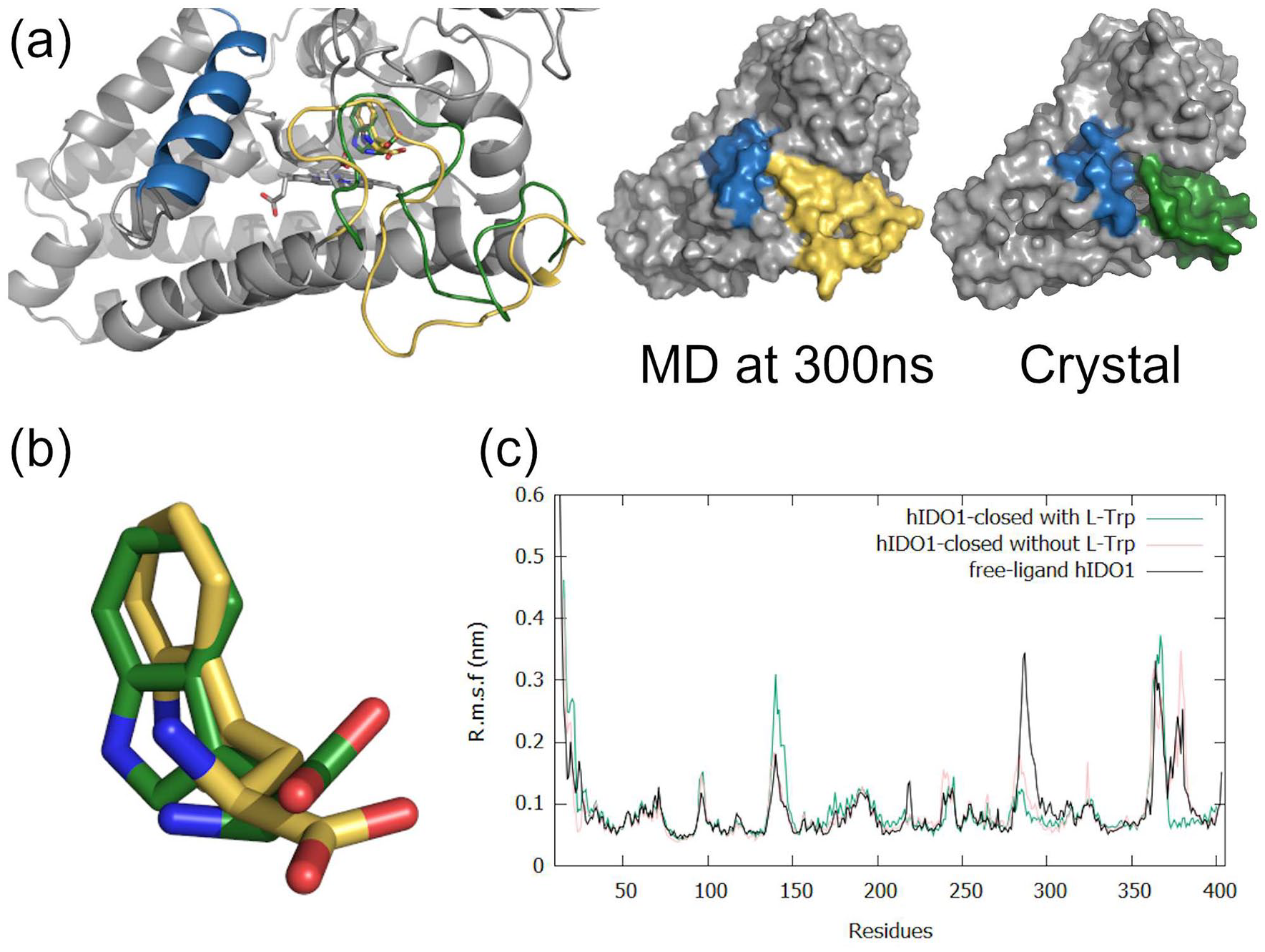
Molecular dynamics studies on hIDO1-closed as obtained from 300 ns of MD simulation at 310 K and 1 bar. (a) Final 300 ns-MD conformation of the JK-loop compared to the hIDO1-closed crystal structure. Induced fit of the JK-loop in hIDO1-closed during the MD production stage in comparison to the crystal structure, respectively in green and yellow while the helix F is in blue. (b) Superposition of the L-Trp conformation in the hIDO1-closed crystal structure (in green) and as obtained from 300 ns of MD simulation (in yellow). (c) RMSF analyses of hIDO1-closed (with and without ligand) in comparison to free-ligand hIDO1 MD studies. 39
In the same way as the 300 ns-MD simulation with L-Trp in the closed conformation, 2 other 300 ns-MD simulations were performed with structures with the JK-loop in intermediate and opened conformations as starting points. First, the 300 ns-MD trajectory for hIDO1 with the JK-loop in the intermediate conformation shows a total reorganization of the loop and ligand position, leading to a drastic increase of the RMSF values for the C-terminal part of the loop (Figure 5a). The L-Trp stays relatively mobile until it finds its definite position within the active site (Figure 5b and c). The more advanced position in the active site is considered as an additional intermediate position of L-Trp in the active site (Figure 5c, in yellow) as is not the known reactive position for the substrate (Supplemental Figure SI4). This position is stabilized after 200 ns until the end of the simulation. During its migration, the distance between the protonated amine of L-Trp and the heme cofactor varies with an average of 3.8 ± 1.3 Å. The displacement of L-Trp leads to the displacement of the JK-loop. The access to the active site of the protein gradually closes. In this intermediate conformation of the JK-loop, K377 points outwards the enzyme. However, the cofactor is maintained in the active site due to the establishment of an interaction between K373 and the substrate acting as a network between the JK-loop, the substrate and the heme (Figure 5c, left). The simulation starting from the reconstructed hIDO1-opened structure does not show the same displacement trend due to the fact that L-Trp does not undergo a full 180° rotation around its initial position. The unproper orientation of the indole ring with respect to the heme cofactor and the closure of the pocket A prevent the displacement of L-Trp in the active site. Consequently, L-Trp remains in its position and the JK-loop is still open. In a biological context, it is hypothesized that L-Trp would potentially slightly exit the enzyme to rotate and to re-enter in the correct orientation.

Molecular dynamics snapshots as obtained from a 300 ns MD simulation at 310 K and 1 bar with hIDO1-intermediate as the starting point: (a) RMSF analyses of hIDO1-intermediate in comparison to free-ligand hIDO1 and hIDO1-closed simulations, (b) Position of L-Trp at 1 ns of production (in red) in comparison to the position observed in the crystal structure (in orange), and (c) Left—2 MD snapshots of the displacement for the JK-loop during the production stage after 1 ns (in red) and after 300 ns (in yellow). Right - Interaction between L-Trp and K373 after 300 ns in yellow. Loss of contact between the heme and K377.
A comparison of the 3 crystal structures, superimposed to MD illustrating transient intermediate positions of L-Trp allows to propose a possible refolding trajectory for the JK-loop. When L-Trp enters in the active site of hIDO1, the loop is first open (Figure 6a, I). L-Trp adopts a temporary intermediate position which corresponds to position 1 or position 2 (Figure 6b) in crystallography. In the case of position 1, it must first perform a 180° rotation to allow a better anchoring of the substrate in the active site to shift in the A-pocket. As a result, the loop changes from an open to an intermediate conformation (Figure 6a, II and III). This conformation is close to the conformation of the loop in the ligand-free structure (Supplemental Figure SI5). Stabilizing interactions are likely established between the loop, L-Trp and the cofactor to allow the confinement of the heme and the substrate into the active site (Figure 6c). The substrate is displaced into pocket A, through a movement from position 2 to position 3 and then 4 (Figure 6b) through favorable hydrophobic stabilizations. The stabilization of the L-Trp in this final position allows the closure of the active site (Figure 6a, IV). If a dioxygen molecule is previously bound to the iron ion under the right oxidation-redox conditions, the reaction can take place. After reaction, it is suggested that less favorable interactions with the product of the reaction lead to the opening of the loop and the release of the product.

Superposition of the different conformations for the JK-loop in the 3 PDB structures and the MD simulations. The structure hIDO1-closed is in green, hIDO1-intermediate in orange and hIDO1-opened in blue. Structures from MD simulation after 300 ns is in yellow: (a) JK-loop opening according to the different structures, (b) trajectory of L-Trp in the different structures, and (c) interactions network between the JK-loop, the substrate and the heme to maintain the cofactor in the active site in the MD simulation and in hIDO1-closed structure.
Influence of the crystallographic conformations on the affinity
All the above results indicate the occurrence of a strong interaction between the heme cofactor and the C-terminal part of the JK-loop. In order to evaluate the resulting affinity, MM-GBSA calculations were performed (Table 1). Concerning the holo protein/substrate complexes, the results highlight the same order of affinity for the different crystal positions of L-Trp with respect to the holo protein. In all cases, these negative values for the binding ∆G for the 3 structures support the possible observation of these temporary intermediates in crystallography. It suggests that the influence of the conformation of the loop on the substrate affinity is negligible. Contrarily, the affinity of the heme for the protein is strongly influenced by the conformation of the loop. In the open-loop situation, the binding ∆G is positive, showing no affinity of the heme for the enzyme. Thus, such a conformation state favors the apo versus the holo form. When the loop is in an intermediate position (hIDO1-ligand free and hIDO1-intermediate with L-Trp), the binding ∆G for the cofactor presents a negative value which favors the holo rather than the apo form. It can be explained by the electrostatic interaction observed between K377 and the cofactor. The closure of the enzyme drastically decreases the ∆G by a factor of 10, leading to an increase of the heme affinity and, consequently, a decrease of the heme lability. This analysis supports the role of the JK-loop on the modulation of cofactor lability.
MM-GBSA calculations of the different PDB structures with a partially or totally refined JK-loop.

Influence of the JK-loop conformation on the affinity for L-Trp or heme cofactor. The affinity for L-Trp or heme cofactor is calculated on the basis of the position as observed in the corresponding crystal structure. In the case of the affinity of the heme cofactor, the substrate is removed from the active site during the calculation. Values of ∆G are in kcal/mol.
Questioning the existence of an exo site in hIDO1
In the 3 crystal structures, it is possible to observe L-Trp or NFK molecules outside the enzyme, mostly at the interface between monomers in the crystal packing (Supplemental Figure SI6). However, in the 3 crystal structures reported in the present work, L-Trp and NFK molecules are observed in some monomers, with no other monomer less than 10 Å away by crystal symmetry. Such a location site, called as exo site, is located in the small subunit (I50-R56 and W92-R100). Within the structure hIDO1-closed, L-Trp is found in monomer B. In hIDO1-intermediate, 2 monomers (B and C) contain L-Trp and NFK, respectively. The hIDO1-open structure contains an NFK molecule in monomer A. In addition, MD analyses of a free NFK outside the protein lead to its displacement around the exo site during the 300 ns MD production stage (Supplemental Figure SI7).
L-Trp and NFK are not in the same orientation in the different monomers, as illustrated in Figure 7. It is explained by the amount of electrically charged residues of the exo site, which stabilize the polar groups of the molecules. Nevertheless, the common aromatic moiety of the ligands occupies a pocket (pocket I, in green, Figure 7a) composed of residues L49, I50, G53, and L55. However, the stabilization of the polar part of the ligands can be achieved with 2 other pockets (II and III). Pocket II (Figure 7a, I, in blue) is composed of the residues R100, E57, and/or E60, while the pocket III (Figure 7a, II, in pink) includes the main chain of residues K94 and G93, as well as the lateral chain of D98. The contribution of R56 appears in both polar pockets. The interaction maps (Figure 7b) highlight that the occupancy of pocket II or III is not a ligand-dependent process. Indeed, the position of L-Trp in the hIDO1-closed structure (Figure 7b, I) is close to that of NFK in monomer C of hIDO1-intermediate (Figure 7b, III). In contrast, the polar part of L-Trp in the monomer B of hIDO1-intermediate (Figure 7b, II) is closer to the position of NFK in hIDO1-opened (Figure 7b, IV).

L-Trp and NFK binding in the exo site of the small subunit of hIDO1. Pocket I is in green while pocket II and III are respectively in blue and in pink. (a) I, NFK in monomer C of hIDO1-intermediate as positioned in the pockets I and II. II, L-Trp in monomer B of hIDO1-intermediate as positioned in the pocket I and III. (b) Interaction maps of L-Trp or NFK in the exo site of the different monomers (I = Monomer B of hIDO1-closed, II = Monomer B of hIDO1-intermediate, III = Monomer C of hIDO1-intermediate, IV = Monomer A of hIDO1-opened).
Beside the crystallographic and MD results, a computational study was performed by Maestro 46 to determine the possible binding sites at the protein surface. The calculations were performed using the SiteMap tool on monomers with or without a ligand in the exo site. Although the known active site of hIDO1 is recognized as the best site with a site score higher than 1.0, 46 the program also finds the site of the small subunit with a lower score around 0.6 to 0.7 (Table 2). The volume of the active site is lower in the case of monomer A in hIDO1-closed due to the closed conformation of the JK-loop. In the 2 other monomers, it is more open due to the absence of refinement of the JK-loop. According to this approach, the exo site has an overall size between 45 and 90 Å3 (Table 2). This shows some variability of the pocket depending on the monomer. Surprisingly, the hIDO1-intermediate structure is characterized by narrower binding sites. So far, it is not possible to establish a link between the position of the loop and the size of the exo site. Observation of the site surface interacting with L-Trp or NFK inside shows that this exo site is indeed able to accommodate an NFK as well as a L-Trp molecule (Supplemental Figure SI8). All these results support the recent study performed by Greco et al 40 who suggested that an exo site may be present in hIDO1. The study of the role for this site is an interesting track to follow in the near future.
SiteMap analyses of for the different monomers with ligand in the exo-site or in the active site in the 3 hIDO1 crystal structures.

Conclusions
The dynamic loop of hIDO1 has long been a poorly understood area in the enzyme. To shed light on the reaction pathway of the protein, we present results obtained through X-ray diffraction experiments, molecular dynamics (MD) simulations, MM-GBSA calculations and protein binding site analysis. In particular, 3 new crystal structures highlight 3 possible conformations of the JK-loop. These conformations can be related to both (1) the entry and position of the ligand in the active site and (2) the binding of the cofactor inside the enzyme pocket involving the residue K377. MD trajectories have supported the existence of these conformations of the JK-loop in solution and revealed additional snapshots at the 3 temporary intermediates observed in crystallography. The set of these 5 positions allowed a better understanding of the dynamics of the JK-loop during substrate positioning. Together with the MM-GBSA results, it is now clear that the JK-loop has a fundamental role in the lability of the heme cofactor. All the results form a solid basis for the design of holo or apo inhibitors for the protein. The analysis of the crystal structure, as well as protein binding site analysis, confirm the previously published hypothesis of Greco et al 40 about a novel exo site in hIDO1, which remains a challenging and unexplored road. The precise characterization of the exo site is crucial to understand its role within the small subunit of the enzyme.
Supplemental Material
sj-pdf-1-try-10.1177_11786469211052964 – Supplemental material for Temporary Intermediates of L-Trp Along the Reaction Pathway of Human Indoleamine 2,3-Dioxygenase 1 and Identification of an Exo Site
Supplemental material, sj-pdf-1-try-10.1177_11786469211052964 for Temporary Intermediates of L-Trp Along the Reaction Pathway of Human Indoleamine 2,3-Dioxygenase 1 and Identification of an Exo Site by Manon Mirgaux, Laurence Leherte and Johan Wouters in International Journal of Tryptophan Research
Footnotes
Acknowledgements
The authors thank the Soleil Synchrotron and the PROXIMA-1 and PROXIMA-2 beamlines staffs for the access to synchrotron-radiation facilities and for the precious advices during the experiment. The authors are also grateful to Professor Liang Tong for providing the hIDO1 plasmid. This research used the resources of the Plateforme Technologique de Calcul Intensif (PTCI; http://www.ptci.unamur.be) located at the University of Namur, Belgium, which is supported by the FNRS-FRFC, theWalloon Region and the University of Namur (Conventions Nos. 2.5020.11, GEQU.G006.15, 1610468, and RW/GEQ2016). The PTCI is a member of the Consortium des Equipements de Calcul Intensif (CECI). (![]() ).
).
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: MM acknowledges the Fonds de la Recherche Scientifique (F.R.S.-FNRS, Belgium) for her Research Fellow grant.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
The authors’ contributions are as follows. As head of the laboratory, JW designed the project and supervised the work. For the experimental part, MM performed the experiments, refined and analyzed the different crystallographic structures with the feedback of JW. For the theoretical part, the Molecular Dynamics protocols were developed by MM and LL. The interpretation of the results has been done in consultation with the different authors of the present manuscript. During the writing process, MM took the lead of the redaction. All authors provided critical feedback and helped the analysis of the manuscript..
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.