
Abstract
Modulation of tryptophan (Trp) metabolism may underpin the behavioral effects of androgenic-anabolic steroids (AAS) and associated image and performance enhancers. Euphoria, arousal, and decreased anxiety observed with moderate use and exercise may involve enhanced cerebral serotonin synthesis and function by increased release of albumin-bound Trp and estrogen-mediated liver Trp 2,3-dioxygenase (TDO) inhibition and enhancement of serotonin function. Aggression, anxiety, depression, personality disorders, and psychosis, observed on withdrawal of AAS or with use of large doses, can be caused by decreased serotonin synthesis due to TDO induction on withdrawal, excess Trp inhibiting the 2 enzymes of serotonin synthesis, and increased cerebral levels of neuroactive kynurenines. Exercise and excessive protein and branched-chain amino acid intakes may aggravate the effects of large AAS dosage. The hypothesis is testable in humans and experimental animals by measuring parameters of Trp metabolism and disposition and related metabolic processes.
Introduction
Androgenic-anabolic steroids (AAS) are used as image and performance enhancers. They cause positive and negative behavioral effects and addiction potential. The negative effects of high-dosage use or after withdrawal drive users to harmful misuse of addictive drugs and, along with needle injections, have led to significant public health and social concerns. The behavioral effects are likely to be mediated by biopsychosocial determinants; understanding and dissecting their roles are daunting tasks. Psychological theories have been proposed, some (controversial) information is available on potential biochemical determinants. While recognizing their complexity, I would like to hypothesize here a biochemical basis of the behavioral effects of AAS and other enhancers involving changes in metabolism and disposition of the essential amino acid
AAS and enhancers
Androgenic-anabolic steroids are derived from testosterone (T). Three main classes are recognized based on structures of the derivative (Table 1). Class I includes T esters, which are administered intramuscularly. Class II comprises 19 nortestosterone (nandrolone) derivatives, which are also administered intramuscularly. Class III comprises derivatives produced by alkylation at C17 and are taken orally. Testosterone is rapidly metabolized and undergoes 4 types of transformations: (1) irreversible reduction by 5α-reductase to 5α-dihydrotestosterone (5α-DHT); (2) 3α-hydroxylation in brain and reproductive tract to 3α-hydroxylated isomers; (3) oxidation of the 17β-hydroxyl group to the 17β-keto metabolite androstenedione, further successive reductions of which lead to formation of the urinary metabolites androsterone and etiocholanolone; (4) aromatization of T and androstenedione, mainly in adipocytes and brain, to estradiol (E2) and estrone, respectively. To prevent the loss of T’s anabolic activity because of its rapid metabolism, more stable derivatives are produced by esterification of the 17-hydroxyl group or by substitution of the methyl group at C19 with a hydrogen, followed by esterification. Alkylation at the 17α-carbon produces orally effective anabolic derivatives. Other compounds are used orally, although they are not 17α-alkylated, eg, methenolone (metenolone) acetate. Aromatization is limited to classes I and II.
Classes of androgenic-anabolic steroids.

Intake of AAS follows 4 separate patterns: cycling (use of cycles interrupted by a drug-free period, to minimize side effects); stacking (use of more than 1 AAS, to maximize the gain in muscle mass); pyramidal (building up to a maximum dose and then tapering down to rebalance the endocrine system); and continuous intake.
Adjunctive enhancers used include human growth hormone (HGH), human chorionic gonadotropin, insulin, and insulinlike growth factor 1 (IGF-1). Human growth hormone increases muscle and tendon strength. Insulin promotes protein synthesis and increases muscle uptake of amino acids, whereas IGF-1 mediates the effects of HGH, enhances protein synthesis, and inhibits protein breakdown. Human chorionic gonadotropin stimulates production of T by mimicking the action of luteinizing hormone. Other (secondary) agents are used mainly to counteract the side effects of AAS and include diuretics, lipolytic agents, a synthetic thyroid hormone, and an antiestrogen.
Side effects
Side effects vary with the AAS used and include acne, gastrointestinal disturbances, gynecomastia, headache, hypertension, liver damage, premature balding, and water retention. With insulin, there is a risk of hypoglycemia, whereas with HGH, bone overgrowth, cardiomegaly, and a tendency to develop diabetes are likely. 1
Behavioral effects
Positive behavioral effects include euphoria, arousal, and decreased anxiety, whereas negative ones include aggression, anxiety, depression, personality disorders, and psychosis. The behavioral effects of use and withdrawal of these agents and their psychological determinants are discussed in the excellent reviews by Rashid et al 1 and Riem and Hursey. 2 An underlying theme is the difficulty in interpreting the behavioral effects, partly on methodologic grounds and the significant role of expectancies by users. Most previous behavioral studies have methodologic limitations, including differences in dosages, measurements at single time points, rather than longitudinally, interval between chronic use and withdrawal, absence of and the difficulty in choosing the best placebo group, differences in baseline characteristics and, in particular, sensitivity to behavioral effects as influenced by personality traits, and impact of exercise. It is generally thought that low-dose AAS intake is associated with the positive effects, whereas high doses may be linked to the negative ones. Even here, expectancy can have a major impact on the behavioral responses. Assessing the role of expectancy is not an easy task. 2
Overview of Tryptophan Metabolism and Disposition
Introduction
In addition to its essential role in protein synthesis, Trp is the precursor of many important biologically active metabolites (Table 2). These include the mood-controlling cerebral indolylamine serotonin (5-HT), the pineal hormone melatonin involved in the daily rhythm and other important processes, the pellagra-preventing factor niacin (nicotinic acid or nicotinamide: vitamin B3), the important redox cofactors nicotinamide adenine dinucleotide (phosphate) (NAD+(P+)) and their reduced forms reduced nicotinamide adenine dinucleotide (phosphate) (NAD(P)H), the neuroactive kynurenine (Kyn), and its various metabolites, namely, the N-methyl-
The tryptophan metabolic pathways.

The tryptophan metabolic pathways
As outlined in Table 2, Trp is metabolized by 4 pathways, 3 of which are of minor quantitative importance. These are the hydroxylation, decarboxylation, and transamination. The fourth, the oxidation or kynurenine pathway (KP), is the quantitatively most important, accounting for ~95% of overall dietary Trp disposal.3,4 The KP exists largely in the liver, wherein it accounts for ~90% of Trp oxidation under normal physiological conditions. The KP also exists extrahepatically, but its contribution to Trp oxidation is normally small (5%-10%), but can assume greater significance under conditions of immune activation, with important biological consequences. 5
Because the KP controls Trp metabolism (see below), it is important to describe features of particular relevance to the present hypothesis. A detailed review of the regulatory and functional aspects of the KP has recently been published, 5 and various accounts of Trp metabolism are freely available to the nonexpert from this journal’s Web site. The KP is controlled by the first enzyme: Trp 2,3-dioxygenase (TDO) in liver and indoleamine 2,3-dioxygenase (IDO) elsewhere. Both TDO and IDO are hemoproteins. Trp 2,3-dioxygenase in livers of humans and some other animal species exists in 2 forms: the active holoenzyme and the inactive heme-free apoenzyme, which becomes activated by heme. Trp 2,3-dioxygenase activity can therefore be inhibited by inactivation of the holoenzyme or prevention of conjugation of the apoenzyme with heme. Indoleamine 2,3-dioxygenase exists in the active form that cannot be further activated by heme. Trp 2,3-dioxygenase is upregulated by 3 mechanisms: hormonal induction by glucocorticoids and some other hormones, substrate activation and stabilization by Trp, and cofactor activation by heme. 5 Indoleamine 2,3-dioxygenase is upregulated by IFN-γ and other cytokines acting via IFN-γ and downregulated by nitric oxide (NO). Thus, activity of the recombinant human enzyme is reversibly inhibited by NO binding to heme, with the inactivated enzyme complex being the Fe2+-NO-Trp adduct. Inhibition may provide a mechanism of regulating the immune function of IDO (for reference, see Badawy 5 ). Trp 2,3-dioxygenase has a high capacity and low affinity for Trp, whereas the opposite is true for IDO, whose activity is inhibited by high concentrations of Trp (Ki > 200 µM). 5
The cerebral serotonin pathway is primarily dependent of Trp availability to the brain. Of the 2 enzymes of 5-HT biosynthesis (Figure 1), Trp hydroxylase (TPH) is rate limiting. Tryptophan hydroxylase exists largely (at least 50%) unsaturated with its Trp substrate.
6
Accordingly, moderate changes in brain [Trp] can cause significant changes in 5-HT synthesis. Tryptophan hydroxylase activity can be influenced by other factors and undergoes substrate inhibition7,8 which may be of particular importance in the present hypothesis. Although the second step in 5-HT synthesis, catalyzed by aromatic
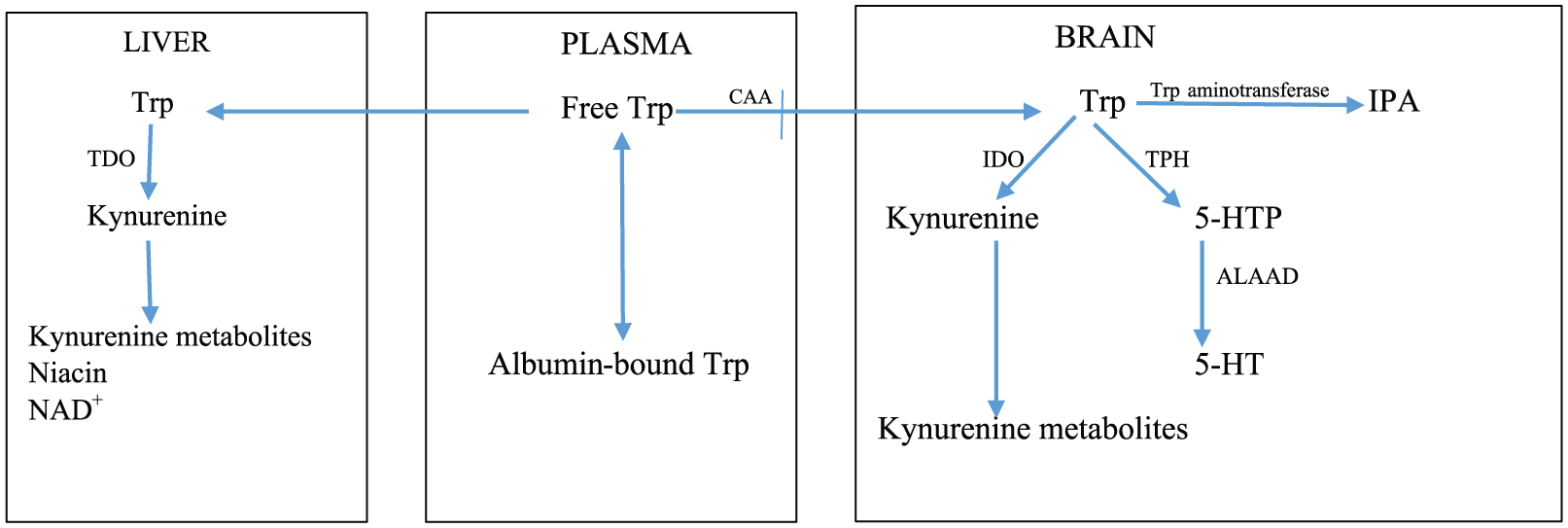
Tryptophan disposition between the liver and the brain. 5-HT indicates 5-hydroxytryptamine; 5-HTP, 5-hydroxytryptophan; ALAAD, aromatic
Brain Trp can also undergo transamination to indolepyruvic acid (IPA) via an unstable intermediate. As will be discussed below, IPA possesses anxiolytic properties and can also be converted to one of the intermediates of the KP, KA.
Control of plasma tryptophan availability by the KP
The KP plays a major role in control of plasma Trp availability for cerebral 5-HT synthesis and other processes. Studies in mice from which the TDO and IDO genes were deleted have established the major role of liver TDO under normal physiological conditions in control of plasma Trp concentration and availability to the brain for serotonin synthesis.10–13 Although cerebral uptake of plasma Trp is determined primarily by liver TDO activity, at the secondary and more immediate level, uptake is determined by plasma Trp binding to albumin and extent of competition with Trp from a number of competing amino acids (CAA) that share the same carrier mechanism, mainly the 3 branched-chain amino acids (BCAA) leucine, isoleucine, and valine and the 2 aromatic amino acids phenylalanine and tyrosine (Figure 1). In human studies, changes in brain [Trp] can best be predicted from changes in the ratio of plasma [Trp]/[CAA].
Plasma tryptophan disposition
Plasma Trp exists largely (90%-95%) bound to albumin, with the remaining 5% to 10% being free. The flux of Trp down the KP and the uptake of Trp in brain and other organs are primarily the functions of free Trp. 5 However, equilibration between the free and the albumin-bound fractions is so rapid that a sustained increase in free [Trp] and the consequent increase in its tissue uptake can result in depletion of the bound fraction. It is therefore important that both free and total (free + albumin-bound) Trp are measured. This can help establish not only the baseline status of Trp metabolism and its physiological determinants but also the interpretation of changes induced by exogenous factors or disease states. 14 Tryptophan binding is usually expressed as the percentage of free Trp (100 × [Free Trp]/[Total Trp]).
As regards Trp entry into the brain, the role of free Trp has been the subject of some controversy during the 1970s-1980s, which can be resolved as follows. Although it is true, as stated above, that free (non–albumin-bound) Trp faces competition from CAA from dietary sources15,16 (see below), it can enter the brain both directly while freely circulating and also after being released from albumin-binding sites after enhanced dissociation at the blood-brain barrier (BBB). This is because the affinity of the BBB to Trp is greater than that of albumin.17,18
Plasma-free Trp is a labile parameter easily influenced by a variety of physiological and pharmacological factors, 14 such as the physiological binder albumin and the physiological displacers, nonesterified fatty acids (NEFA), and some hormones and drugs, all of which act by altering albumin binding. Table 3 lists the various conditions under which plasma-free Trp can be altered and the mechanisms involved. As will be described below, some of these conditions are met with use of AAS and by exercise.
Factors affecting plasma-free tryptophan concentration.

Abbreviations: IDO, indoleamine 2,3-dioxygenase; TDO, tryptophan 2,3-dioxygenase.
↑ indicates increase, − indicates no change, ↓ indicates decrease,? denotes a potential effect of testosterone.
Food can also influence plasma-free [Trp], with a high-fat or high-protein meal increasing it and a high-carbohydrate meal decreasing it. However, Trp availability to the brain for 5-HT synthesis does not reflect these patterns, at least with protein and carbohydrate. Thus, the increase in free [Trp] following a high-protein meal actually leads to decreased entry of Trp into the brain because of increased competition for cerebral uptake from the CAA, as levels of the latter in proteins are much greater than that of Trp. 15 By contrast, the decrease in free [Trp] after a carbohydrate meal, which is mediated by an insulin-dependent inhibition of lipolysis, is associated with increased entry of Trp into the brain because by stimulating muscle uptake of amino acids, insulin undermines the competition with Trp from CAA.15,16 With fat diets, the increase in serum-free [Trp] is reflected in elevation of brain [Trp] and enhanced 5-HT synthesis. 19 These changes may be involved in influencing Trp disposition through the dietary habits of athletes.
Although liver TDO is the major determinant of Trp availability for brain 5-HT synthesis, the latter can also be impaired if brain IDO is induced by IFN-γ under conditions of immune activation and brain injury. Here, cerebral Trp metabolism is diverted toward the KP and away from the serotonin pathway. Indoleamine 2,3-dioxygenase induction in brain (or periphery) leads to increased formation of kynurenine and its metabolites, many of which are neuroactive. In particular, KA and QA are, respectively, the antagonist and agonist of the NMDA subtype of receptors of the excitatory amino acid glutamate. 20 Kynurenic acid is therefore cytoprotective, with, among others, anxiolytic properties, whereas QA is cytotoxic, inducing hyperexcitability, anxiety, and neuronal damage.
Although cerebral uptake of KA and QA is limited, that of kynurenine is relatively much greater. 21 This suggests that cerebral synthesis of KA and QA could be enhanced by a rise in circulating [Kyn]. However, as will be stated below, a significant proportion of brain QA is of peripheral origin and impaired BBB permeability under certain conditions could result in increased brain entry of QA and also KA. In the above study, 21 albumin binding in rat serum showed that, although QA does not bind, 27% of Kyn and 71% of KA exist in the bound forms. The circulating concentration of Kyn is the highest among kynurenines and the question of whether Kyn and Trp can influence cerebral uptake of one another through competition at albumin-binding sites or for the neutral amino acid L carrier system has been the subject of investigation and critical review. 21 Briefly, only very high concentrations of Kyn (1 mM) that are unachievable in vivo under physiological or pathological conditions would be required to inhibit Trp transport into the brain. From kinetic data in rats, Fukui et al 21 calculated that the apparent Km for Kyn transport into the brain is some 2000-fold higher than the physiological [Kyn], and a 10-fold increase in plasma [Kyn] (which can occur after acute Trp loading in humans)22–26 can only exert a negligible effect on neutral amino acid transport. Evidence to the contrary, however, exists. Thus, Gál et al 27 reported that a 5 mg/kg intraperitoneal dose of Kyn inhibits Trp uptake by various rat brain structures. By contrast, Trp can inhibit Kyn transport into the brain. At a 10 mM concentration, Trp causes a 95% inhibition of Kyn uptake by rat mouse astrocytes. 28 At equimolar concentrations of Trp and Kyn (30 µM), Kyn uptake is inhibited by Trp by 40%. 28 As plasma [Trp] under basal conditions and after acute or chronic loading is several orders of magnitude greater than [Kyn], competition from the latter is unlikely to occur.
The Hypothesis and its Background
The hypothesis consists broadly of the following statement. Androgenic-anabolic steroids and other enhancers exert positive effects on image and behavior when used in low or moderate doses and in conjunction with exercise but cause negative effects when used excessively and also on their abrupt withdrawal. Both the positive and the negative effects on behavior involve modulation of Trp metabolism in the appropriate direction: enhanced mood, on the one hand, and lowered mood, increased anxiety, and aggressive behavior, on the other hand. Increased serotonin may underpin the enhanced mood, whereas the lowered mood, anxiety, and aggression may be mediated by lowered serotonin and increased production of psychoactive kynurenine metabolite. Other neurochemical correlates of these behavioral changes have been proposed in the literature and will be discussed later on.
The proposed hypothesis encompasses broadly the changes listed in Table 4. The impact of exercise on the above processes could influence the behavioral changes. The following is a discussion of evidence in support of these potential effects.
Proposed mechanisms of the behavioral effects of AAS and other enhancers.

Abbreviations: 5-HT, 5-hydroxytryptamine or serotonin; AAS, androgenic-anabolic steroids, E2, estradiol; GH, growth hormone; IDO, indoleamine 2,3-dioxygenase; IPA, indolepyruvic acid; KA, kynurenic acid; PLP, pyridoxal phosphate; QA, quinolinic acid; T, testosterone; TDO, tryptophan 2,3-dioxygenase; TPH, tryptophan hydroxylase; Trp,
Possible displacement of albumin-bound tryptophan by testosterone
Testosterone binds to sex hormone–binding globulin (SHBG) and albumin. In men and women, albumin binds 54% and 20% of circulating T, respectively. 29 Another study 30 reported that, in men, albumin binds 53% to 55% and SHBG binds 43% to 45%. In women of fertile age, the corresponding values are 36% to 37% and 62%, respectively. Thus, the binding capacity of T to SHBG is lower in men than in women, 30 although the association constant is greater in men. There are, however, no sex differences in these binding parameters for albumin. 31 The free unbound fraction of T is 2% in men and 1.5% in women. 30 Estradiol binding was also calculated. 30 In men and women, the free E2 fraction is 2.4% and 2.0% of the total, respectively. In men, albumin binds 68% to 70% and SHBG binds 28% to 30% of E2. In women, the corresponding values are 52% and 45% to 46%. Although it is generally believed that it is the free fraction of biologically active molecules that determines tissue uptake, with T and E2, both the free and the bound fractions are equally important, but this depends on the tissue. 32 Thus, albumin-bound T is mostly freely transported into brain and liver but only partially into salivary gland and lymph node, whereas SHBG-bound T is not transported into tissues. By contrast, SHBG-bound E2 is transported into liver, salivary gland, and lymph node, but not into brain, whereas albumin-bound E2 is transported into the brain. 32 Tissue-mediated enhanced dissociation is responsible for these differences. 32 The effects of weekly injections of various doses of T enanthate on plasma total and free [T] were studied in younger and older men. 33 Significant dose-dependent increases in both total and free [T] were observed with the weekly doses of 125, 300, and 600 mg. The smaller doses of 25 and 50 mg were not associated with elevated [T]. Compared with baseline values, the above 3 larger doses resulted in rises in total [T] of 2.20-fold, 5.72-fold, and 9.08-fold, respectively. The corresponding rises for free [T] were 2.22-fold, 6.94-fold, and 12.82-fold, respectively. This suggests that T-binding sites on albumin are saturated by doses of 125 mg and above. Sex hormone–binding globulin binding is not involved in the increase in free [T], as all doses of the androgen caused a nonsignificant 12% decrease in this binding protein.
There are 2 possible mechanisms by which T may increase plasma-free [Trp] by displacement from albumin-binding sites. The first is direct displacement by T, whose affinity to human serum albumin (6.6 × 10−4 M) 34 is relatively greater than that of Trp (9.7 × 10−3 M). 35 The second is indirect displacement via NEFA. The latter is known to be elevated by T in the goldfish Carassius auratus. 36 As far as I could ascertain, no studies have been published on a potential effect of T on plasma Trp binding in humans or other mammalian species.
Conversion testosterone to E2
Conversion, which has been demonstrated in rat 37 and human 38 ovary slices in vitro, has desirable physiological consequences across animal species, ranging from acceleration of gonadal differentiation and stimulation of spermatogenesis in prepubertal sea bass 39 and modulation of respiratory long-term facilitation in male rats 40 to neuroprotection in humans. 41 The effects of dose and age on the conversion of injected T to E2 were studied in humans in detail. 42 At baseline, compared with older men (age range: 60-75 years), younger men (age range: 18-35 years) exhibit higher serum-free and total (free + protein bound) T and lower total, but not free, E2. Calculated as the percentage of free T, older men have a 33% greater T binding, which corresponds to their 32% higher SHBG concentration. When subjects received weekly injections of T for 23 weeks, dose-dependent increases in serum-free and total E2 were observed, which were significantly greater in older men. A previous study by the same group established that the serum-free and total T elevation after T administration is greater in older men. 33
A potential explanation of the greater elevation of E2 in older men is that aromatase activity increases with age. Aromatase (also known as estrogen synthase) is a member of the cytochrome P450 family of enzymes (P450arom), a product of the CYP19 gene, that catalyzes the aromatization of androgens to estrogens using O2 as cosubstrate and NAD(P)H as cofactor. Thus, it converts T to E2 and androstenedione to estrone. Aromatase has a wide tissue distribution. That aromatase activity is increased by aging has been demonstrated in adipose tissue of postmenopausal women43,44 and older men.45,46 However, irrespective of age, the above findings and the gynecomastia caused by performance and image enhancers confirm the conversion of AAS to estrogens. It should, however, be pointed out that aromatization of AAS to estrogens is limited to classes I and II and not class III.
Inhibition of liver TDO activity by estrogens
It is currently accepted that estrogens inhibit liver TDO activity and impair hepatic Trp metabolism in rats and humans, thereby increasing Trp availability to the brain, despite earlier reports of TDO induction. Trp 2,3-dioxygenase induction by E2 was blocked by adrenalectomy or actinomycin D, thus suggesting that it is mediated by or requiring the presence of glucocorticoids.47–50 Subsequent studies in rats, mice, and humans, however, clearly established the TDO inhibitory effects of E2 and also progesterone. Thus, TDO activity of rat liver homogenates is inhibited by administration of E2 or progesterone by a mechanism involving inactivation of the holoenzyme and prevention of conjugation of the apoenzyme with heme. 51 Elevation of E2 and progesterone may also explain the TDO inhibition during the first 15 days of rat pregnancy. 51 When TDO activity is assayed in rats in vivo, ie, by measuring the production of 14CO2 from ring 2- 14 C-Trp, E2 administration decreases the release of 14CO2. 52 Tryptophan 2,3-dioxygenase inhibition is further suggested by the finding that the increase in rat plasma [Trp] following an acute Trp load is further enhanced by E2 administration. 52 An increase in serum-free and total [Trp] in humans also occurs by administration of estrone or the synthetic estrogen ethinyl estradiol. 53 The percentage of free Trp (an expression of Trp binding to albumin) is not altered by these 2 estrogens (13.5 and 14.4, respectively compared with a control percentage of 13.4) As outlined in Table 3, these Trp elevations in the absence of altered Trp binding typify TDO inhibition and, in fact, ethinyl estradiol, which induces destruction of hepatic heme, impairs cytochrome P450–dependent drug metabolism, 54 and also inhibits TDO activity (unpublished work referred to in Morgan et al 55 ). The decreased urinary excretion of kynurenine and the lower rise in plasma kynurenine after acute Trp loading observed in women receiving oral contraception (ethinyl estradiol plus progestogens) 56 further support TDO inhibition by estrogens.
Effects of estrogens on tryptophan availability to the brain
A consequence of TDO inhibition by E2 is an increase in circulating Trp availability to the brain for central serotonin synthesis. Surprisingly, little work has been done in this regard. A small decrease in cortical [Trp] in female, and in mid-brain [5-HT] in male, rats has been reported. 48 In rat pregnancy, a sustained increase in concentrations of brain Trp, 5-HT, and 5-HIAA during the period of TDO inhibition occurs, 51 which can be attributed to both estrogens and progesterone. Estrone administered in the rat diet at a concentration of 0.002% (w/w) exerts no effect on serum or brain Trp nor on brain 5-HT and 5-HIAA. 57 However, as stated above, 53 a smaller dose of estrone (1.5 mg twice daily) given to menopausal women increases both free and total plasma [Trp]. Other indirect evidence for elevated brain 5-HT synthesis secondarily to increased Trp availability in humans is provided by the findings that plasma total [Trp] and the [total Trp]/[CAA] ratio are increased in females using ethinyl estradiol and also to a lesser extent its 3-methyl ether mestranol 58 and the correlation between plasma estrogens and free Trp, which was suggested to involve direct displacement by estrogens from albumin-binding sites. 59 As will be discussed below, estrogens can influence brain serotonin function in other ways.
The question arises as to whether the extent T conversion to E2 is sufficient to induce a TDO inhibition and a consequent elevation of plasma [Trp]. A 600-mg T dose per week increases plasma [E2] by 2.7-fold in younger men (from 76 to 202 pM) and by 3.5-fold in older men (from 96 to 338 pM). 33 Such elevations are within the normal range of total E2 in women during the follicular phase and exceed that in the luteal phase. As some users of AAS can take more than thrice the above T dose daily, it is very likely that even larger amounts of E2 will be formed to inhibit TDO activity and increase Trp availability to the brain for 5-HT synthesis.
Effects of estrogens on brain serotonin synthesis and function
Estrogens can also influence cerebral serotonin synthesis and function in other ways. Thus, in macaques, (1) ovariectomy decreases serotonin neuron numbers and gene expression 60 ; (2) E2 stimulates the formation of the TPH protein through induction of its messenger RNA (mRNA), 61 although this would still require an adequate supply of the Trp substrate because, as stated above, TPH exists partially (~50%) saturated with Trp; (3) ovarian steroids protect serotonin neurons 62 ; and (4) exogenous E2 induces an anxiolytic response in rats in association with increased TPH protein and serotonin transporter levels,63,64 with estrogen receptor β regulating the expression of the TPH2 mRNA. 65
The relationship between E2 and serotonin synthesis from its immediate precursor 5-hydroxytryptophan (5-HTP) is less clear. The PLP-dependent brain ALAAD activity and the 5-HT content in 6-hour postmortem human samples are considerably lower than those in experimental animals. 9 The effects of E2 or ethinyl estradiol on activities of various aminotransferases or their saturation with the PLP cofactor in experimental animals are controversial, most likely due to species-, organ- and subcellular-specific differences and type of sex steroid.66–70 For example, whereas E2 and ethinyl estradiol may inhibit, progestogens stimulate these enzymes. In humans, this is illustrated by the absence of an effect of a combination of norgestrel and ethinyl estradiol on alanine aminotransferase or aspartate aminotransferase. 71 It is thought that the estrogen inhibition of PLP-dependent enzymes does not involve depletion of PLP. It is therefore unlikely that E2 will influence ALAAD activity or availability of its PLP cofactor. Only one study in mice examined sex differences in activity, mRNA expression, and protein content of ALAAD acting on 3,4-dihydroxyphenylalanine (Dopa). 72 Sex differences were observed only in kidney and small intestine. The above 3 ALAAD parameters were higher in female kidney compared with males, whereas in intestine, males had higher values. These differences appear to be a function of androgens and may be related to sex differences in sodium balance. Thus, there is no evidence that estrogens impair ALAAD. This is further supported indirectly by the generally well-demonstrated enhancement of 5-HT function by estrogens.
Effects of HGH, IGF-1, and insulin
The relationship between GH and IGF-1 typifies a negative feedback mechanism. Thus, whereas GH secretion is inhibited by IGF-1, synthesis of the latter is enhanced by GH. As the major anabolic hormone, GH acts by switching fuel consumption from proteins and carbohydrates to lipids. This is achieved by GH-activating GH receptors and enhancing synthesis of IGF-1, which stimulates protein synthesis and inhibits protein degradation, and by directly enhancing lipolysis and the release of NEFA. 73 The lipolytic action of GH is mediated by the β-3 adrenoceptor, which activates the protein kinase A cascade leading to lipase activation. Through this mechanism, GH enhances lipolysis and the lipolytic effect of catecholamines in adipocytes and lipoprotein lipase activity in skeletal muscle, thereby facilitating NEFA utilization. 74 This may be of particular relevance to the present hypothesis. Enhanced lipolysis leading to an increase in circulating NEFA is the most sensitive response to GH. 75 Infusion of GH at a rate of 45 ng/kg/min doubles plasma NEFA. 76 For a 70-kg adult, this corresponds to 3.15 µg/min. As body builders/image enhancers self-administer doses of GH between 2 and 32 IU, 77 corresponding to 0.66-10.56 mg, a considerable increase in [NEFA] can be expected under these conditions.
Insulinlike growth factor 1 exerts both GH-like and insulinlike actions that are dependent on dosage, length of treatment, and route of administration. 77 The 2 main actions of IGF-1 are suppression of GH secretion and increased muscle utilization of NEFA. Although at physiological concentrations IGF-1 does not stimulate lipolysis, its administration to GH-deficient subjects enhances lipolysis (by suppressing insulin secretion) and also NEFA utilization by muscles. 77
Insulin is antilipolytic. It inhibits the breakdown of triglycerides (triacylglycerols) to NEFA and strongly enhances lipogenesis. Although this causes decreased free [Trp], overall, the effect of insulin is to enhance Trp entry into the brain, as stated above, by increasing muscle uptake of amino acids, thus undermining the competition from CAA with Trp for cerebral uptake.
Testosterone, other enhancers, cortisol, and the brain
Testosterone and other enhancers can influence cortisol release with potential implications for brain function, including serotonin synthesis. Cortisol inhibits cerebral 5-HT synthesis by decreasing Trp availability to the brain via liver TDO induction.
Testosterone suppresses cortisol release at the adrenal level. Thus, it decreases significantly corticotropin-releasing hormone (CRH)-stimulated cortisol levels and peak cortisol, although paradoxically it increases CRH-stimulated ACTH (adrenocorticotrophic hormone; corticotropin) secretion. 78 In this study, 78 male subjects were in the age range of 18 to 45 years and received intramuscular injections of 200 mg of T enanthate every 2 weeks for 1 month. A smaller dose of T enanthate (100 mg) given biweekly for 26 weeks to older men (68-73 years) does not influence serum cortisol. 79 Thus, with relatively large T doses, we could expect a decrease in circulating cortisol, which can lower liver TDO activity, thereby increasing Trp availability to the brain for 5-HT synthesis. Age and T dosage are clearly important determinants of this potential effect.
Growth hormone can also influence cortisol, though, in a less clear fashion. In pigs, porcine GH increases transiently serum cortisol but at very high doses (1000 µg/kg of body weight). 80 A smaller parenteral dose of GH in humans (0.1 mg/d) given for 3 months does not influence cortisol metabolism, assessed by measuring the enzyme catalyzing the interconversion of cortisone and cortisol (11β-hydroxysteroid dehydrogenase). 81 In hypophysectomized rats, cortisol induces a greater and longer-lasting induction of liver TDO and also tyrosine aminotransferase than in adrenalectomized or intact rats, and GH administration reverses this effect of hypophysectomy. 82 This suggests that GH can undermine TDO induction by cortisol and hence protect against a consequent cerebral serotonin depletion. However, in contrast, another study 83 reported that hypophysectomy of female rats or dwarf mice is associated with elevated brain [Trp] and the major serotonin metabolite 5-hydroxyindoleacetic acid (5-HIAA) and that these increases are reversed by GH.
Insulin, as stated above, increases brain [Trp] and hence 5-HT synthesis by decreasing the competition from CAA with Trp for entry into the brain by stimulating their muscle uptake, despite lowering plasma-free [Trp] by inhibiting lipolysis. In normal subjects, insulin-induced hypoglycemia stimulates hypothalamic-pituitary-adrenal (HPA) axis activity, resulting in elevation of cortisol and ACTH.84,85 Increased HPA axis activity is higher in type II diabetes, compared with controls, 86 especially in those with diabetic complications/metabolic syndrome.86,87 However, reduction in circulating insulin with benfluorex does not alter plasma cortisol levels. 88
The cortisol status following IGF-1 is more complicated because of the interactions between IGF-1, GH, and insulin (see above). The dosages of these 3 enhancers (GH: 2-32 IU/d; insulin: 2-60 U/d; IGF-1: 20-120 µg/d) 77 render them likely to modulate cortisol levels, although the brain 5-HT status will depend on the balance between the cortisol changes and the T effects.
Testosterone, other enhancers, cytokines, and the brain
Cytokines can influence brain function in 2 ways: enhancing Trp oxidation down the cerebral KP leading to production of neuroactive kynurenine metabolites and depriving the serotonin pathway from its Trp precursor. The outcome will depend on the type of cytokine altered by T and enhancers against the serotonin-promoting effects of T. Neuroactive kynurenine metabolites could also be formed in brain if plasma kynurenine levels are elevated either through TDO/IDO induction or through simply increased flux of Trp down the hepatic KP. Flux of free plasma Trp is the major determinant of kynurenine metabolite formation 5 and as plasma free [Trp] is likely to be elevated by T, kynurenine formation may also be increased, unless TDO is inhibited simultaneously (see below). As far as I could ascertain, there is currently no information on the effects of T or enhancers on plasma kynurenine or its metabolites. Testosterone, however, appears to possess anti-inflammatory and neuroprotective properties, eg, in multiple sclerosis (MS) 89 and intracranial lymphocytic choriomeningitis viral infection. 90 In line with these properties, T lowers the expression and secretion of tumor necrosis factor α (TNF-α) and interleukin 1β (IL-1β), 91 and IFN-γ production. 90 Testosterone, however, does not inhibit IFN-γ induction of IDO in mouse lung. 92 Free serum T shows a negative correlation with the pro-inflammatory IL-6 and pregnancy-associated plasma protein A in patients with acute coronary syndrome. 93 Taken together, this suggests that T is unlikely to induce peripheral or central IDO and consequently will not undermine serotonin synthesis in brain. Whether it can increase plasma and hence brain kynurenine levels to stimulate the production of neuroactive kynurenines remains to be established. It is possible, however, that this may occur if plasma free [Trp] is strongly elevated by large doses of T (see below). It is important to emphasize here that entry into the brain of kynurenine metabolites is rather limited, whereas that of kynurenine itself is not.
Growth hormone does not alter pro- or anti-inflammatory cytokine levels under acute conditions, 94 whereas after intake more than 3 months, it lowers levels of circulating pro-inflammatory cytokines and markers of inflammation, including TNF-α and IL-6 in cardiomyopathy patients. 95 A large GH concentration (100 ng/mL), however, enhances the release of pro-inflammatory cytokines (IL-1β, IL-6, and TNF-α) from lipopolysaccharide-stimulated human monocytes. 96 Thus, excess GH can induce a pro-inflammatory response, leading to IDO induction and a potential increase in neuroactive kynurenine metabolites and a possible central serotonin deficiency.
Although pro-inflammatory cytokines can lower the expression of IGF-1, neuronal death induced by such cytokines (IL-1β or IFN-γ) can be reversed by IGF-1. 97 Thus, IGF-1 exerts a neuroprotective effect and is therefore unlikely to cause harm. However, the potential inhibition of GH secretion by IGF-1 (see above) should be taken into consideration.
Insulin is also unlikely to modulate Trp metabolism negatively along the KP, as it possesses anti-inflammatory properties. For example, it antagonizes the pro-inflammatory effects of hyperglycemia in hospitalized patients 98 and the glucose-induced release of several pro-inflammatory cytokines from normal periodontal ligament cells. 99
Impact of Exercise
As well as influencing levels of cortisol, T and enhancers, exercise can affect many of the effects of T and enhancers at multiple levels. Changes are also influenced by intensity of exercise and its duration (eg, single session or long term).
Effects of exercise on cortisol, T, and enhancers
Exercise increases cortisol, T, GH, and IGF-1 to varying extents depending on experimental conditions,85,100–105 with the cortisol elevation being dependent on the time of day. 106 Whether this cortisol elevation can affect liver TDO activity requires assessment. However, in rats, prolonged exercise increases both circulating corticosterone and liver TDO activity. 107 Exercise, however, lowers insulin. 108 Thus, the effects of exogenous T, GH, and IGF-1 are likely to be potentiated by exercise.
Other effects of exercise
These have recently been reviewed 109 and will only be summarized below. As the T elevation appears to be confined mainly to free T, 104 it is possible that this could reflect displacement from albumin-binding sites by NEFA. That exercise elevates circulating NEFA is a well-documented effect, resulting from enhanced lipolysis secondarily to increased release of adrenal medullary catecholamines. 110 The NEFA elevation results in displacement of Trp from its albumin-bound sites, leading to a free Trp elevation, an effect observed in both humans and experimental animals. 109 An increase in Trp availability to the brain can be expected to increase brain [Trp] and hence serotonin synthesis. This has been demonstrated in rats. 111 In humans, the increase in the [Free Trp]/[CAA] ratio is due to the free Trp elevation and not a decrease in [CAA]. 109 The increases in [NEFA] and [Free Trp] induced by T are therefore likely to be potentiated by exercise. However, if large doses of T are used, an excessive elevation of free Trp may lead to undesirable behavioral effects (see below).
As stated above, liver TDO activity is enhanced by exercise in rats in parallel with the corticosterone elevation. 107 The doubling of circulating cortisol in humans 109 suggests that TDO activity may also be enhanced. However, it is possible that this enhancement may be countered if sufficient E2 is produced from classes I and II AAS. Nevertheless, a TDO enhancement in the presence of a free Trp elevation could increase the chances of a raised plasma [Kyn], as has been demonstrated in humans and rats.112–114 An increase in plasma kynurenine can enhance cerebral kynurenine levels leading to production of neuroactive kynurenine metabolites with potential behavioral consequences. Blood QA is also increased by exercise and it has been suggested that QA can cross the BBB, 115 which is weakened by exercise. 116 Tumor necrosis factor α, whose serum levels are increased by exercise,112,115 has previously been shown to impair BBB permeability.117,118 Furthermore, 40% to 50% of brain QA is of peripheral origin. 119 Thus, although exercise may be beneficial in conjunction with moderate doses of AAS, it may contribute to the undesirable behavioral effects of large doses. In the above study in rats, 114 enhancement of macrophage IDO activity was reported, as was the elevation of the pro-inflammatory cytokine TNF-α in the human study. 112 In fact, various pro- and anti-inflammatory cytokines are elevated by exercise (for references, see Badawy 109 ), and the net effect on IDO activity can be determined by the balance between these 2 types of cytokines and also by their interactions.
As stated above, exercise increases plasma [Kyn], and it has been reported 120 that endurance exercise induces a transient increase in plasma [KA] by activating skeletal muscle kynurenine aminotransferase (KAT) and that this may involve activation of the PGC-1alpha1 gene, which has earlier been demonstrated by the same group in mice. 121 The authors120,121 suggested that KAT induction diverts Kyn metabolism toward the neuroprotective KA and away from the neurotoxic 3-HK and QA and that this renders KAT induction a potential strategy for treatment of stress and depression. However, the results of these 2 studies require further scrutiny and may be subject to other interpretations. Thus, first, as KAT activity was not measured in either study, it remains to establish whether the expressed KAT proteins are catalytically active. For example, the considerable increases in the mRNA and protein expressions of 3 KATs did not result in decreases in plasma [Kyn] or increases in plasma [KA]. 121 Second, assuming the increased KATs are catalytically active, they will still require sufficient amounts of the Kyn substrate to activate them, given the high Km of KATs for Kyn. 5 An increase in [Kyn] is therefore a critical prerequisite for KAT activation. In the human study, 120 plasma [Kyn] was not measured, whereas in the mouse study, 121 it was elevated by mild chronic stress. Third, transgenic mice with skeletal muscle-specific PGC-1alpha1 (mck-PGC-1α1) overexpression do not differ from the wild type in any of the KP parameters measured in plasma, liver, or brain. The TDO/IDO induction by chronic mild stress can explain the elevation of plasma [Kyn]. Rats exposed to a variety of stressors (eg, cold exposure) have been shown122,123 to develop a new glucocorticoid receptor specifically associated with induction of liver TDO but not another glucocorticoid-inducible enzyme, tyrosine aminotransferase. Liver TDO induction by cortisol or activation by catecholamines leading to a central serotonin deficiency may be an important feature of major depressive disorder, 124 and studies in mice 10 showed that deletion of the TDO gene results in decreased anxiety. A rise in plasma [Kyn] in these mice was not associated with changes in [KA], and the authors 10 concluded that the anxiolytic effect of TDO gene deletion is attributable to enhanced cerebral serotonin synthesis and turnover. Fourth, although TDO induction can lead to decreased serotonin synthesis and increased Kyn-derived KA, it is important to ascertain whether mck-PGC-1alpha1 overexpression protects against anxiety caused by chronic mild stress by blocking TDO/IDO induction, enhancing KAT’s catalytic activity, or both. Finally, measuring plasma-free and total [Trp] under the above conditions could have been informative. In particular, as stress induces a catecholamine-dependent lipolysis-mediated release of albumin-bound Trp 14 and a glucocorticoid-mediated TDO induction,122,123 conditions could have been created to optimize the increases in plasma [Kyn] through TDO/IDO induction superimposed on increased free [Trp], especially as the latter is the major determinant of the Trp flux down the KP. 5 Superimposed on exercise, a greater increase in plasma [Kyn] is likely to occur with chronic use of class III AAS, than with classes I and II, as the latter 2 classes may undermine TDO induction by exercise through a potential action of estrogens.
Previous Studies of Neurochemical Correlates of the Behavioral Changes
The positive effects of AAS and enhancers (arousal and reward, lack of anxiety, and enhanced mood) are likely to be experienced with use of moderate doses and in conjunction with exercise, whereas the negative effects (anxiety, irritability, dysphoria, depression, aggression, personality disorders, and psychosis) could occur with use of large supraphysiological or “mega” doses, long-term intake, and on withdrawal. Many excellent reviews and original investigations of the neurochemical correlates of these behaviors, mostly in experimental animals, have been published. Findings in many cases are controversial, being dependent on sex, species, and drug. Moreover, attributing a specific behavior to a single neurochemical is difficult because of multiple interactions at the neurophysiological level. A summary of these studies is given below, in which an attempt will be made to relate the findings to the present hypothesis. In their comprehensive review of animal studies, Clark and Henderson 125 concluded that T is anxiolytic but enhances aggressive behavior, whereas nandrolone does not exert these effects. Stanozolol, by contrast, lowers aggression. This may reflect the inability of the latter to be converted into estrogens. Androgenic-anabolic steroids appear not to influence learning memory or to modify reward directly, although they may potentiate the rewarding effects of amphetamine: a potentiation that reflects possible sensitization by AAS of the dopamine-dependent reward circuitry, which may explain why AAS abusers may abuse other drugs. 125 Large doses of AAS are capable of influencing many neuronal systems. Thus, they activate androgen receptors and increase their immunoreactivity, allosterically modulate GABAA receptors, lower serotonergic tone, and alter the expression of dopamine and its receptors.
That T may play a role in major depressive disorder is suggested by the observation 126 in older men of a significantly lower free and total T in such patients, compared with controls. As regards psychosis, a number of observations also implicate T as a positive modulator. For example, T influences excitement/hostility levels 127 and is negatively correlated with the severity of negative symptoms in schizophrenia. 128 Testosterone and other AAS exert various important effects on NMDA receptor subunits.129–132 Of particular importance are findings related to excitotoxicity mediated by activation of NMDA receptors. Thus, nanomolar concentrations of some AAS (nandrolone, stanozolol, and gestrinone) amplify NMDA toxicity in mixed mouse cortical cultures. 133 By contrast, T amplifies neuronal cell death only at very high concentrations (10 µM) but is protective at 10 nM, with intermediate concentrations being without effect. 133 A situation could be visualized wherein NMDA receptor activation induced by the kynurenine metabolite QA can interact with high levels of AAS to induce excitotoxic responses, eg, anxiety and psychosis. In a widely used mouse model of MS (experimental autoimmune encephalomyelitis), which involves among others a faster decay rate of NMDA receptor–mediated currents, T preserves excitatory synaptic neurotransmission in the hippocampus. 134 It is of interest that in a patient with MS, cerebrospinal fluid (CSF) [QA] is elevated several-fold (from a normal range of 10-35 to 168 nM). 135 Quinolinic acid is also elevated in spinal cord of rats with experimental allergic encephalopathy. 136 The QA elevation appears to reflect the disease stage and progression. In the above study, 135 the CSF QA of a patient with MS was measured during an acute attack. Quinolinic acid elevation in the CSF of patients with MS occurs in relapsing-remitting patients in relapse and in primary progressive patients, 137 reflecting activation of the KP. 138 As stated above, another harmful situation could arise with use of high doses of AAS (especially class III AAS) in conjunction with exercise.
Withdrawal of (usually large doses of) AAS results in a withdrawal syndrome not dissimilar to that experienced by drug-dependent patients. Symptoms include anxiety, irritability, insomnia, hot flashes, sweats, chills, anorexia, myalgia, nausea, vomiting, tachycardia, and hypertension. 139 Depression and craving may also occur. Anxiety could in part be related to a serotonin deficit, as suggested by the findings in adolescent hamsters of decreased serotonin innervation in brain areas implicated in anxiety and the ability of the specific serotonin-reuptake inhibitor (SSRI) fluoxetine to reduce withdrawal anxiety. 140 In this latter study, the AAS doses were not relatively very excessive (2 mg/kg each of T and nandrolone and 1 mg/kg of DHT daily for 30 consecutive days). As far as I could ascertain, no information on plasma Trp disposition during AAS withdrawal is available. However, a hypothesis could be proposed. If liver TDO is inhibited by estrogens produced from classes I and II AAS, abrupt withdrawal may cause a rebound induction of TDO causing a decrease in Trp availability to the brain and a consequent inhibition of serotonin synthesis. Such a rebound TDO induction and impairment of cerebral serotonin synthesis, induced by an increased release of corticosterone, occur on withdrawal after TDO inhibition by chronic administration to rats of diverse agents: glucose, nicotinamide, 141 and 4 different drugs of dependence. 142 Liver TDO is a target of a large number of antidepressant drugs. 124 A role of estrogens in AAS withdrawal is further suggested by several observations: TDO inhibition, antidepressant effects in postnatal depression, acceleration of the antidepressant effects of SSRIs, and occurrence of a withdrawal syndrome on their abrupt withdrawal (for reference, see Badawy 124 ).
Another important issue is induction of aggressive behavior by large doses of T and other AAS. A paradox arises here in reconciling the expected elevation of plasma-free and brain [Trp] and 5-HT synthesis and the well-documented role of serotonin dysfunction in aggression. Although most of the studies associate a low brain serotonin synthesis and turnover with aggressive behavior,143–147 a higher plasma [Trp] in aggressive subjects has been reported by some.148–150 In one study, 150 free and total serum [Trp] and [CAA] were all elevated without a significant change in the [Free Trp] / [CAA] or [Total Trp]/[CAA] ratios. The proportionate increases in free and total Trp (68% and 64%, respectively) suggest a TDO inhibition. However, with TDO inhibition, the Trp elevations are usually ~25%. 142 Other explanations are possible based on whether aggression still involves a serotonin dysfunction or another mechanism. An even stronger serotonin deficiency could still occur with excess Trp acting by 2 mechanisms: substrate inhibition of TPH7,8,151 and PLP depletion through Trp transamination to IPA and inactivation by estrogen metabolites. In one study in rats, 151 brain [5-HT] and [5-HIAA] were elevated by doses of Trp of 10 and 25 mg/kg of body weight but started to decline with larger doses, whereas plasma and brain [Trp] continued to rise dose-dependently throughout the full dose range, as did concentrations of kynurenine and 3-HK. Another mechanism may involve increased production of cerebral neuroactive kynurenine metabolites. This can occur if availability of plasma kynurenine is increased or if cerebral IDO is induced preferably with increased Trp availability to the brain. Plasma kynurenine is actually decreased in aggressive subjects.152,153 Regarding AAS, it is possible that class III steroids, which do not undergo aromatization, may allow Trp conversion to kynurenine to proceed uninhibited. Cerebral IDO induction will occur if pro-inflammatory cytokine levels are raised in brain. Studies of cytokines in aggression have only recently begun. Plasma IFN-γ is not altered, whereas IL-10 is only slightly decreased, with the IFN-γ/IL-10 ratio showing a significant correlation with aggression scores. 154 A potential increase in pro-inflammatory cytokines in the brain is therefore more likely with AAS, but, in the absence of evidence for such an increase, the most likely explanation of AAS-induced aggression is a serotonin deficiency caused by the excess Trp. The dissociation of aggression from psychosis regarding cytokine changes 154 further suggests a mechanism not involving KA or QA.
Testing the Hypothesis and Further Comments
From the above discussion, it is clear that the net effects of AAS use will depend, among others, on type and dosage of AAS and of enhancers and their interactions and the impact of exercise. We cannot be certain of exact levels of changes but it is reasonable to attempt here, as outlined in Table 5, to predict the likely changes and suggest laboratory means of testing them. Although some of these likely changes are based on current information, others are predictions based on what is known. The hypothesis is therefore amenable to experimental scrutiny in both human and experimental studies.
Proposed effects of testosterone and other enhancers on parameters of tryptophan metabolism.
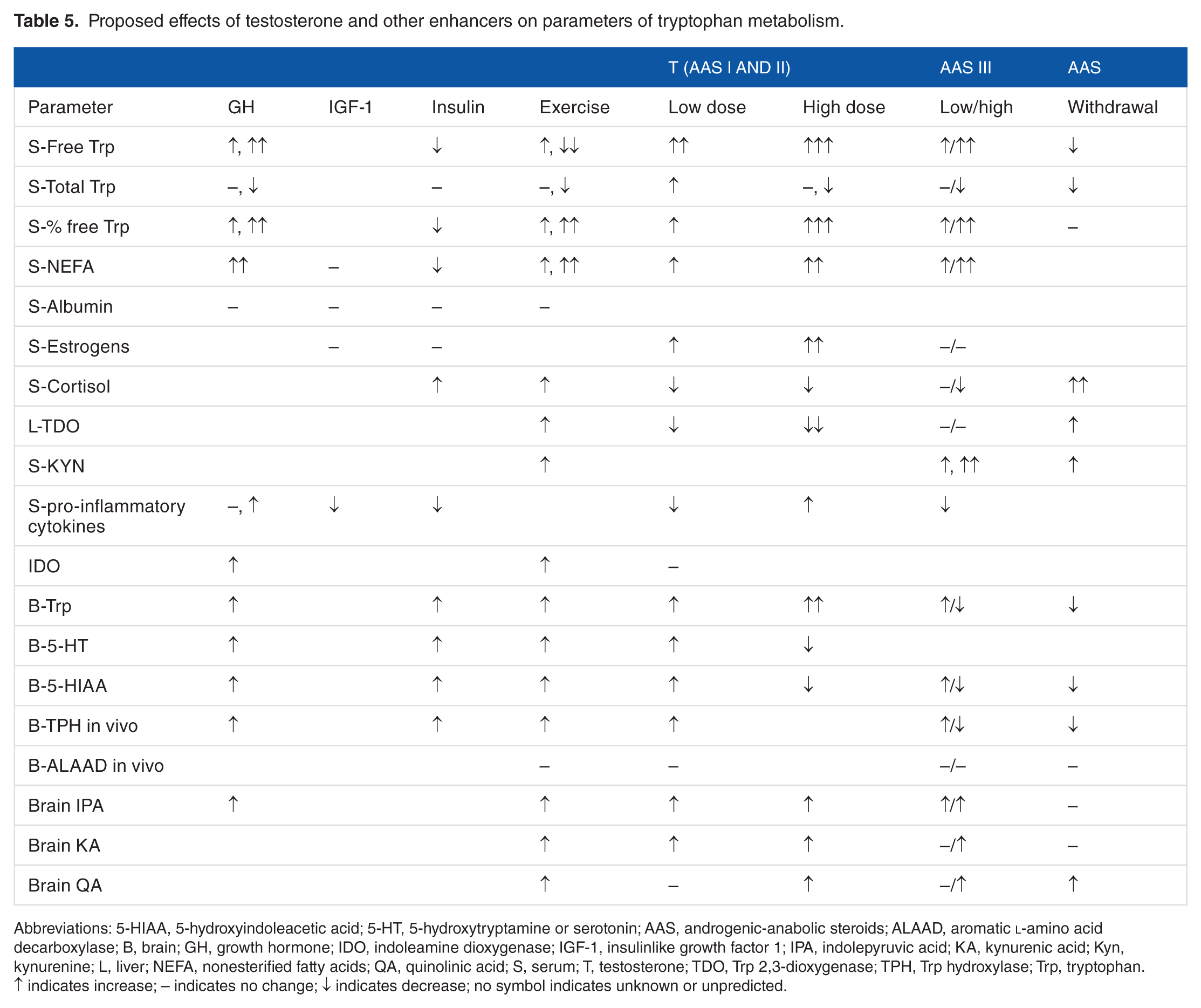
Abbreviations: 5-HIAA, 5-hydroxyindoleacetic acid; 5-HT, 5-hydroxytryptamine or serotonin; AAS, androgenic-anabolic steroids; ALAAD, aromatic
↑ indicates increase; – indicates no change; ↓ indicates decrease; no symbol indicates unknown or unpredicted.
The following is a brief summary statement of the elements of the hypothesis derived from the contents of Tables 3 and 5 and the text:
Use of moderate doses of AAS and enhancers along with exercise should maximize the benefits. Enhanced mood, decreased anxiety, and stimulated reward should result. There should be increases in concentrations of plasma-free Trp and possibly also total Trp (with classes I and II AAS), NEFA, and estrogens and decreases in serum cortisol and pro-inflammatory cytokines and liver TDO activity with moderate AAS use. Serum Kyn is likely to be increased by class III more than classes I and II AAS. In brain, increases can be expected in concentrations of Trp, 5-HT, 5-HIAA, KA, IPA, and possibly also QA. The increases in free Trp and NEFA are likely to be potentiated by exercise and growth hormone but undermined by insulin. However, the increases in brain indoles could be potentiated by both GH and insulin. The potential effects of IGF-1 cannot be predicted, except for its ability to inhibit GH secretion and consequently its likely effects on Trp and other related parameters in the absence of concomitant GH intake.
Use of large doses of classes I and II AAS is likely to be associated with negative behavioral changes. We can expect greater increases in plasma-free and possibly also total Trp and in estrogen levels and a decrease in cortisol resulting in liver TDO inhibition. Brain Trp could be greatly increased resulting in elevation of brain IPA, KA, and QA, as serotonin synthesis may be inhibited by the excess Trp. The enhanced synthesis of KA and QA will be the combined result of increased Trp availability, conversion of IPA to KA, and IDO induction by pro-inflammatory cytokines. The changes in plasma Trp disposition and serotonin synthesis could be partially mitigated by moderate doses of GH or insulin and by exercise, except that the latter could potentiate the rise in QA. Neuronal hyperexcitability and damage could occur with large doses of class II AAS under these conditions. The negative symptoms experienced with use of high-dosage AAS will depend, among others, on potential modulation by exercise and enhancers.
The effects of use of class III AAS will depend on dosage. Moderate doses will influence Trp metabolism and disposition in a broadly similar manner to classes I and II, and concomitant use of GH or insulin and also exercise may exert a potentiating influence. However, because of their inability to be converted into estrogens, class III AAS will not inhibit liver TDO activity and thus will allow the flux of Trp down the KP to proceed. A plasma Kyn elevation would result. With large doses, the plasma Trp and Kyn changes will be exaggerated but serotonin synthesis will be undermined. Growth hormone, insulin, or exercise may mitigate these effects. However, as is the case with class II AAS, neuronal hyperexcitability and damage could occur if QA levels are elevated and exercise may be a contributory factor.
Withdrawal of AAS of various classes resulting in various symptoms is likely to lead to liver TDO induction by cortisol and consequent decreases in plasma Trp and its availability for cerebral serotonin synthesis. The resulting increase in serum [Kyn] can lead to increased production of KA and QA in brain. It is possible that the withdrawal syndrome may be stronger with classes I and II, as it may include aspects specific to estrogen withdrawal (anxiety, irritability, depression, among others), whereas that of class III AAS may include other symptoms related to NMDA receptor activation. The withdrawal syndrome is more likely to be stronger or more persistent if enhancers such as GH or insulin are withdrawn simultaneously.
The role of exercise during the AAS withdrawal phases deserves comment. It is generally the case that AAS users are unlikely to exercise if having severe withdrawal symptoms, which may necessitate hospitalization. Exercise energy is likely to be impaired during withdrawal. 155 However, anecdotal evidence suggests that exercise (and also low-dose AAS or human chorionic gonadotropin administration) may ameliorate the withdrawal symptoms.2,156
The following are additional comments. Brain TPH activity assayed in suitable tissue preparations is likely to be increased by classes I and II by virtue of their aromatization to estrogens. Assayed in vivo (ie, by measuring brain 5-HTP following ALAAD inhibition by NSD-1015), an increase in TPH activity would be expected with all 3 classes of AAS if brain [Trp] is elevated.
Dietary habits of bodybuilding AAS users could affect the behavioral effects if excessive amounts of proteins are consumed. A decrease in Trp entry into the brain resulting in lowered serotonin synthesis is likely to occur, especially if less carbohydrate is consumed simultaneously. A similar lowering of serotonin will occur if supplements of BCAA are taken, as these will compete with Trp for cerebral uptake. Although these negative effects on brain serotonin could be mitigated, at least partially, by exercise and moderate use of AAS, excessive use of the latter under these conditions is likely to be harmful.
An increase in plasma kynurenine and its metabolites is more likely to occur with class III AAS because of the absence of TDO inhibition by estrogens. With concomitant exercise and excessive protein and BCAA supplemental intakes, the adverse behavioral effects of large doses of AAS are more likely to be greatest with class III compounds followed by class II and least with class I.
In conclusion, it is hoped that the proposed hypothesis will act as a catalyst for further investigation of the behavioral effects of AAS. Although AAS have many clinical applications 157 (eg, in aplastic anemia, cancer, growth retardation, human immunodeficiency virus [HIV] infection, osteoporosis, renal, and hepatic failure), very little information from the many clinical studies can inform the current hypothesis because emphasis in these studies was placed on the end points, eg, body weight in growth retardation, bone marrow stimulation in leukemia or aplastic anemia, and appetite and muscle mass in cancer and HIV infection. As many of these conditions have an important immunologic element, the therapeutic efficacy of T and other AAS suggests that they act at least in part via their anti-inflammatory effects.
Many questions remain unanswered. For example, no studies have specifically examined the negative behavioral effects of each of the 3 AAS classes individually because users often use more than one class. Are the behavioral effects of supraphysiological doses of AAS worsened by concomitant exercise or high dietary intake of protein and BCAA supplements as this hypothesis suggests? Does the withdrawal syndrome occur irrespective of the pattern of AAS intake? Does tapering result in a milder syndrome, if at all? Further research of these and other pertinent questions in conjunction with the proposed hypothesis may help provide answers that can usefully lead to prevention and management of the undesirable consequences of AAS use.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: A.A-B.B. held an honorary professorial appointment at Cardiff Metropolitan University during the period September 2006 to September 2016.
Author Contributions
The author is the sole contributor to this article.
Disclosures and Ethics
The author has provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality, and (where applicable) protection of human and animal research subjects. The author has read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The author has also confirmed that this article is unique and not under consideration or published in any other publication and has permission from rights holders to reproduce any copyrighted material. The external blind peer reviewers report no conflicts of interest.