
Abstract
Indoleamine 2,3-dioxygenase-2 (IDO2) is 1 of the 3 enzymes that can catalyze the first step in the kynurenine pathway of tryptophan metabolism. Of the 2 other enzymes, tryptophan 2,3-dioxygenase is highly expressed in the liver and has a role in tryptophan homeostasis, whereas indoleamine 2,3-dioxygenase-1 (IDO1) expression is induced by inflammatory stimuli. Indoleamine 2,3-dioxygenase-2 is reportedly expressed comparatively narrow, including in liver, kidney, brain, and in certain immune cell types, and it does not appear to contribute significantly to systemic tryptophan catabolism under normal physiological conditions. Here, we report the identification of an alternative splicing pattern, including the use of an alternative first exon, that is conserved in the mouse Ido1 and Ido2 genes. These findings prompted us to assess IDO2 protein expression and enzymatic activity in tissues. Our analysis, undertaken in Ido2 +/+ and Ido2−/− mice using immunohistochemistry and measurement of tryptophan and kynurenine levels, suggested an even more restricted pattern of tissue expression than previously reported. We found IDO2 protein to be expressed in the liver with a perinuclear/nuclear, rather than cytoplasmic, distribution. Consistent with earlier reports, we found Ido2 −/− mice to be phenotypically similar to their Ido2+/+ counterparts regarding levels of tryptophan and kynurenine in the plasma and liver. Our findings suggest a specialized function or regulatory role for IDO2 associated with its particular subcellular localization.
Introduction
A major fate of tryptophan (Trp) is catabolism through the kynurenine (Kyn) pathway. The first reaction in this pathway is catalyzed by tryptophan 2,3-dioxygenase (TDO) or indoleamine 2,3-dioxygenase (IDO) enzymes. Activation of the pathway produces a range of biological effects through the depletion of Trp and the formation of biologically active metabolites.1,2 Mammals possess both TDO and IDO, with a gene duplication of IDO, having given rise to 2 IDO enzymes, indoleamine 2,3-dioxygenase-1 (IDO1) and indoleamine 2,3-dioxygnease-2 (IDO2).3–5 The gene duplication occurred before the divergence of vertebrates; however, many lower vertebrates possess only IDO2, having lost the IDO1 gene, suggesting a conserved ancestral function for the IDO2 gene. 6 Mammalian IDO1 has evolved into an enzyme with high efficiency for Trp metabolism, relative to the IDO2 and TDO enzymes. 6 In addition, mouse IDO1 has a signaling capacity that is independent of its enzymatic activity. 7
Mouse and human IDO2 have the capacity to metabolize Trp; although under in vitro conditions used to assay IDO1, the mouse IDO2 enzyme has lower efficiency than mouse IDO1, and the human IDO2 enzyme has lower efficiency than either.3–5 The low enzymatic efficiency is conserved in IDO2 enzymes throughout vertebrate evolution, at least under assay conditions optimized for IDO1 activity. 6 However, the enzymatic efficiency of mouse 8 and human9,10 IDO2 enzymes can be increased with alternative reaction conditions, suggesting a distinct biochemistry between IDO1 and IDO2 enzymes. Mouse IDO2 has some similarity to both TDO and IDO1 in its expression pattern, but it is not known whether this leads to it performing similar physiological functions. Tryptophan 2,3-dioxygenase is expressed abundantly in the liver, and gene deletion leads to a high Trp concentration in plasma.11,12 Mouse IDO2 also is expressed constitutively in the liver, but its deletion does not affect plasma Kyn levels in mice. 13 Mouse IDO1 is constitutively expressed in some tissues, for example, the epididymis, 14 but not in the liver. IDO1 expression can be induced widely by stimuli, for example, the pro-inflammatory cytokine IFN-γ. IDO1 activity is associated with a suppression of immune responses. 15 IDO1 expression has been detected in liver samples of patients with hepatitis and antigen-presenting cells of rat liver allografts.16,17 Mouse IDO2 expression also can be induced in immune cells and has a role in control of regulatory T lymphocytes, although IDO2 does not act in the same manner as IDO1 in suppressing inflammatory skin carcinogenesis. 13 In another inflammation model in Ido1 and Ido2 null mutant mice, IDO2, but not IDO1, was shown to be involved in the production of autoantibodies and development of autoimmune arthritis. 18 The involvement of IDO2 in the development of autoimmune arthritis has been further demonstrated with neutralizing antibodies. 19
In this study, we have extended our studies into mammalian IDO2 function using genetically deficient mice that have been described previously, 13 investigating subcellular localization of the IDO2 protein and its involvement in normal physiology.
Methods
Mice
Mice were bred in the Medical Foundation Building at the University of Sydney. Ido2−/− mice were generated, as described in the work by Metz et al, 13 and possess a deletion of exon 9/10 in the murine Ido2 gene. Genotyping was performed, as described in the work by Metz et al, 13 by extracting genomic DNA, using an Extract-N-Amp Kit (Sigma-Aldrich, Darnstadt, Germany) from the small piece of tissue obtained by an ear punch. Primers for genotyping are listed in Supplementary Table 1. Mice were housed 2 to 5 animals per cage under a 12-hour light-dark cycle with food and water available ad libitum. All studies were conducted in accordance with the New South Wales legislation governing research with animals. The protocols were approved by the University of Sydney Animal Ethics Committee.
IDO2 protein expression.

Stained nuclei count and average stained nuclear surface area in livers of Ido2+/+ and Ido2−/− mice showed a higher number of stained nuclei and average stained surface area per nuclei (μm2) in Ido2+/+ liver than in the knockout counterpart. n = 3 mice in each group with 4 random fields of view counted from each mouse liver section. Data for each mouse are shown and represent the counts of stained nuclei summed from 4 fields of view and stained nuclear surface area averaged over the fields of view.
For the assessment of protein expression and metabolite levels, tissues were collected from 8-week-old mice. For the study of gene expression in a developmental series, outbred wild-type Quackenbush Swiss mice were mated according to protocols approved by the University of Sydney Animal Ethics Committee. Presence of a vaginal plug was considered 0.5 days post coitus (dpc). Whole embryos were collected at 12.5, 16.5, 17.5, and 18.5 dpc, and neonatal livers from days 1, 2, 3, 7, 14, and 56 postpartum. Tissues were either fixed in 15% v/v neutral-buffered formalin (for immunohistochemistry) or snap-frozen and stored at −80°C (for molecular/metabolite analysis). For the latter, tissues were homogenized using a Polytron Tissue homogenizer (PT 2100) in phosphate-buffered saline (PBS) plus protease inhibitors. For molecular studies, the homogenate was diluted with an equal volume of 2× radioimmunoprecipitation assay (RIPA) buffer (50 mmol/L Tris pH 7.4, 150 mmol/L NaCl, 1 mmol/L EDTA, 1% v/v Triton X-100, 1% w/v sodium deoxycholate, 0.1% w/v sodium dodecyl sulfate, and protease inhibitors) for protein extraction and the remaining homogenate added to 350 μL RNA lysis buffer from the ISOLATE II RNA Mini Kit (Bioline, London, UK).
Gene expression
RNA was extracted using the ISOLATE II RNA Mini Kit (Bioline) according to the manufacturer’s instructions. RNA was quantified using a Nanodrop 2000 (Thermo Scientific, Thermo Fisher Scientific, Waltham, MA, USA) and, for microarray analysis only, RNA also was assessed using an RNA 6000 Nano Assay Chip on an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA) as per the manufacturer’s instructions. For microarray analysis on liver RNA from Ido2+/+ and Ido2−/− mice, samples (n > 5) of each mouse strain were pooled such that each individual mouse contributed an equivalent amount of RNA to the pooled sample. Samples were assayed by the Ramaciotti Centre for Genomics, UNSW, using the Illumina mouse (WG-6) BeadChip array system according to the manufacturer’s instructions. Data were extracted using GenomeStudio with the addition of a Partek plug-in to facilitate the analysis of data on Partek software. Data were analyzed using Partek Genomics Suite 6.6 software to identify differentially expressed genes. As no statistical test could be performed on pooled samples, genes identified as having >2-fold change in expression were verified using quantitative reverse transcription-polymerase chain reaction (RT-qPCR) on the individual samples.
For RT-qPCR, 1 µg of total RNA was reverse-transcribed using random hexamers and a Tetro cDNA Synthesis Kit (Bioline). Polymerase chain reaction amplification was performed in 1× KAPA SYBR Fast Universal qPCR Master Mix with 100 nmol/L primers and the complementary DNA synthesized from the equivalent of 50 ng RNA. Amplification was performed in a Rotor-Gene Q (Qiagen) with 40 cycles of 95°C for 15 seconds followed by 60°C for 45 seconds. Quantification of Ido2 and Tdo was performed by the standard curve method using plasmid to create the standard curve. In addition, the presence of transcripts was visualized by agarose gel electrophoresis. For verification of genes identified in the array analysis, the ΔΔCt method was used with normalization to Ywhaz gene transcript. Specificity of amplification was assessed by melting curve analysis or gel electrophoresis of PCR products. Primers are listed in Supplementary Table 1.
Western blot analysis and immunoprecipitation
Protein homogenates in a final concentration of 1× RIPA buffer were incubated on ice for 30 minutes, after which the samples were spun at 16 000 rcf for 15 minutes. The supernatants were assayed for total protein concentration using a bicinchoninic acid (BCA) protein assay (Pierce, IL, USA) according to the manufacturer’s instructions. For Western blot analysis on total protein, 25 µg of protein per well was assayed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane. For immunoprecipitation, the homogenate was precleared by incubation with Protein A. The equivalent of 1 mg total protein was incubated overnight with 2.5 µg antibody (αIDO2 or isotype control) and 40 µL Protein A. After several washes with cold 1× RIPA buffer, the Protein A was resuspended in loading buffer and analyzed by SDS-PAGE and Western blotting using an IDO2 antibody raised in a different species to the one used for immunoprecipitation.
The antibodies assessed for specificity of IDO2 detection included our custom rabbit polyclonal antibody used in a previous study, 3 2 other custom rabbit polyclonal IDO2 antibodies (kind gifts of Dr Ursula Grohmann from the University of Perugia, Italy, and Professor Yasuko Yamamoto from the University of Kyoto, Japan), a commercial rabbit polyclonal antibody (Santa Cruz SC292212; Santa Cruz Biotechnology, Dallas, TX, USA), and a commercial mouse monoclonal antibody (Santa Cruz SC374159; Santa Cruz Biotechnology). Membranes were blocked with Odyssey Blocking Buffer (LI-COR Biosciences, Lincoln, NE, USA) before being incubated with primary antibodies (0.2 µg/mL) overnight. This was followed by 1-hour incubation in the dark with 1:4000 IRDye-labeled secondary antibody (LI-COR Biosciences). Every incubation was performed in Odyssey Blocking Buffer and was followed by 3 to 5 washes of the membrane in PBS with 0.1% v/v Tween 20. The membrane was scanned on an Odyssey Infrared Imaging System (LI-COR Biosciences).
Immunohistochemistry
Immunohistochemistry was performed on formalin-fixed paraffin-embedded tissue using a mouse monoclonal IDO2 antibody (Santa Cruz SC374159; Santa Cruz Biotechnology) and appropriate isotype control (X0943; Agilent). Sections were deparaffinized and rehydrated according to standard protocols. Antigen retrieval was performed by incubating the slides in 0.2 mol/L citrate buffer pH 6.0 at 90°C to 95°C for 12 minutes. After cooling, the slides were washed in 50 mmol/L Tris-buffered saline with 0.1% v/v Tween 20 (TBS-T) for 2 minutes. All subsequent washes were performed for 2 minutes with TBS-T unless mentioned otherwise. Peroxidase blocking was performed thereafter with 3% v/v hydrogen peroxide in methanol for 5 minutes. After the slides were washed, peptide blocking was performed using Mouse on Mouse (M.O.M.) Blocking Reagent (VectorLabs, Burlingame, CA, USA) for 30 minutes. Slides were then washed twice and incubated with 2.5% v/v normal horse serum (NHS) for 5 minutes. Primary IDO2 antibody at a final concentration of 1 μg/mL, prepared in 2.5% v/v NHS, was added to each section and incubated in a humidified container at 4°C overnight. The next morning, the sections were washed twice before the Vector M.O.M. ImmPRESS Reagent (Vectorlabs) was added to the sections and incubated for 10 minutes. Sections were then washed twice for 5 minutes. Dinitrophenyl (DNP) amplification reagent from the TSA DNP (HRP) System (Perkin Elmer, Waltham, MA, USA) was prepared in a 1:50 v/v dilution. This was added to the sections and incubated for 5 minutes. Sections were washed thrice with agitation before 1:100 v/v anti-DNP-HRP, diluted in TNB blocking reagent (both from the TSA kit), was added prior to incubation for 20 minutes. Sections were washed thrice with agitation before diaminobenzidine substrate (Dako) was added and the sections incubated for 4 to 8 minutes (depending on the tissue type). Hematoxylin staining and dehydration were performed using standard protocols. Stained surface area and nuclei count were quantified using the MetaMorph software (Molecular Devices, Sunnyvale, CA, USA).
Metabolite measurements
Tissue homogenates were centrifuged for 5 minutes at 16 000 rcf and the supernatants transferred to fresh tubes. Some of the supernatant was retained for determining the protein concentration using a BCA assay. Trichloroacetic acid (TCA) was added to a final concentration of 4% v/v, and the samples were vortexed and centrifuged for 15 minutes at 16 000 rcf. The supernatants were neutralized with an equal volume of phosphate buffer pH 7.3 before high-performance liquid chromatography (HPLC) analysis of Trp and Kyn. The HPLC system (Agilent 1100 HPLC System) was equipped with a Luna C18 (2) 250 mm × 4.6 mm 5 µm column (Phenomenex, Torrance, CA, USA). Samples were eluted using a gradient 0% to 40% mobile phase B (80% acetonitrile) over 15 minutes at 0.8 mL/min. Tryptophan was detected by fluorescence (excitation at 285 nm and emission at 365 nm) and Kyn by ultraviolet wavelength at 364 nm. The concentrations of Trp and Kyn were determined by the inclusion of a standard curve and were normalized according to the protein concentration before TCA precipitation.
Kynurenine formation in HEK293T cells, transiently transfected with control plasmids or expression constructs for full-length and truncated mouse IDO1 and IDO2, was measured using a previously described method. 3 Our previous studies with this cell line informed us that Kyn is not significantly further metabolized, and the measurement of Kyn formation alone is sufficient for determining enzymatic activity.
For analysis of the metabolism of other amino acids, including Trp, HEK293T cells were transfected with control plasmid or an expression construct for mouse IDO2. 3 Medium (supplemented with some amino acids to assist with detection) was changed 24 hours following transfection and the cell supernatants were collected 24, 30, and 48 hours later. Trichloroacetic acid was added to a final concentration of 10% and the samples centrifuged at 16 000 rcf for 5 minutes. Supernatants were diluted 10-fold in water before analysis. Amino acids were assayed using a modification of Fu et al. 20 First, amino acids were derivatized with o-phthaldialdehyde (OPA) before being separated by HPLC (Agilent 1100) using a OPA-High Speed Column (100 mm × 4.6 mm; Dr. Maisch HPLC GmbH, Ammerbuch, Germany). A gradient of methanol in 20 mmol/L sodium acetate and tetrahydrofuran was used for elution. The flow rate of solvents was set to 1 mL/min, and the excitation and emission was measured by the fluorescence detector at 340 and 440 nm, respectively. To identify the elution profile of the resultant spectrum, samples were spiked with individual amino acids (100-200 µmol/L).
Results and Discussion
The Ido2 gene generates alternatively spliced Ido2 messenger RNA transcripts
Detection of Ido2 messenger RNA (mRNA) is complicated by the existence of alternatively spliced transcripts, including those encoding a variant that is under the regulation of another promoter region. Figure 1A is a schematic diagram showing the position of the exons within the Ido2 gene and the alternatively spliced transcripts. NM_154949 is the transcript encoding the full-length IDO2 protein or NP_666061. XM_006509048 and XM_006509047 were predicted by Gnomon, and we have confirmed their expression in brain RNA using 5′ RACE (SF1). These 2 variants do not contain exon 1, which has the start codon for the full-length IDO2 protein. There is, however, a downstream start codon in exon 3 that could potentially be used to generate a truncated protein, although there is no conclusive evidence to suggest that the variant transcripts are translated into protein. The existence of mRNA variants affects the interpretation of mRNA expression studies as reverse transcription-polymerase chain reaction (RT-PCR) assays using primers spanning exons 1 and 2 show a different pattern of expression compared with primers recognizing either the alternative transcript or the 3′ end of the Ido2 mRNA transcript. The transcript encoding the full-length IDO2 protein revealed highest expression in the liver, followed by the kidney (Figure 1B). In contrast, the brain only expressed the transcript containing the alternative exon 1A (Figure 1B). Primers annealing to Ido2 mRNA further downstream of exon 3 (for example, P5 and P6 in Figure 2B) show expression in all 3 tissues (data not shown). An earlier study also showed more widespread expression of the 3′ end of the mouse Ido2 transcript compared with the full-length transcript, although the alternative first exon was not identified. 4 Interestingly, this alternative splicing pattern also exists for the mouse Ido1 gene, where transcripts with different first exons show tissue-specific expression, presumably due to their different promoter regions (SF2). We examined whether the transcripts of Ido1 and Ido2 with the alternative first exons would generate an enzymatically active protein by cloning the respective open reading frames into an expression vector. Expression of truncated proteins was detected by Western blot analysis (data not shown), but the Kyn levels were no higher than in control-transfected cells (Figure 1B and SF2). This suggests that even if the alternative transcripts seen in the brain and other tissues (XM_006509048, XM_006509047, and NM_001293690) were translated into proteins, they would have little or no enzymatic activity in standard assay conditions.
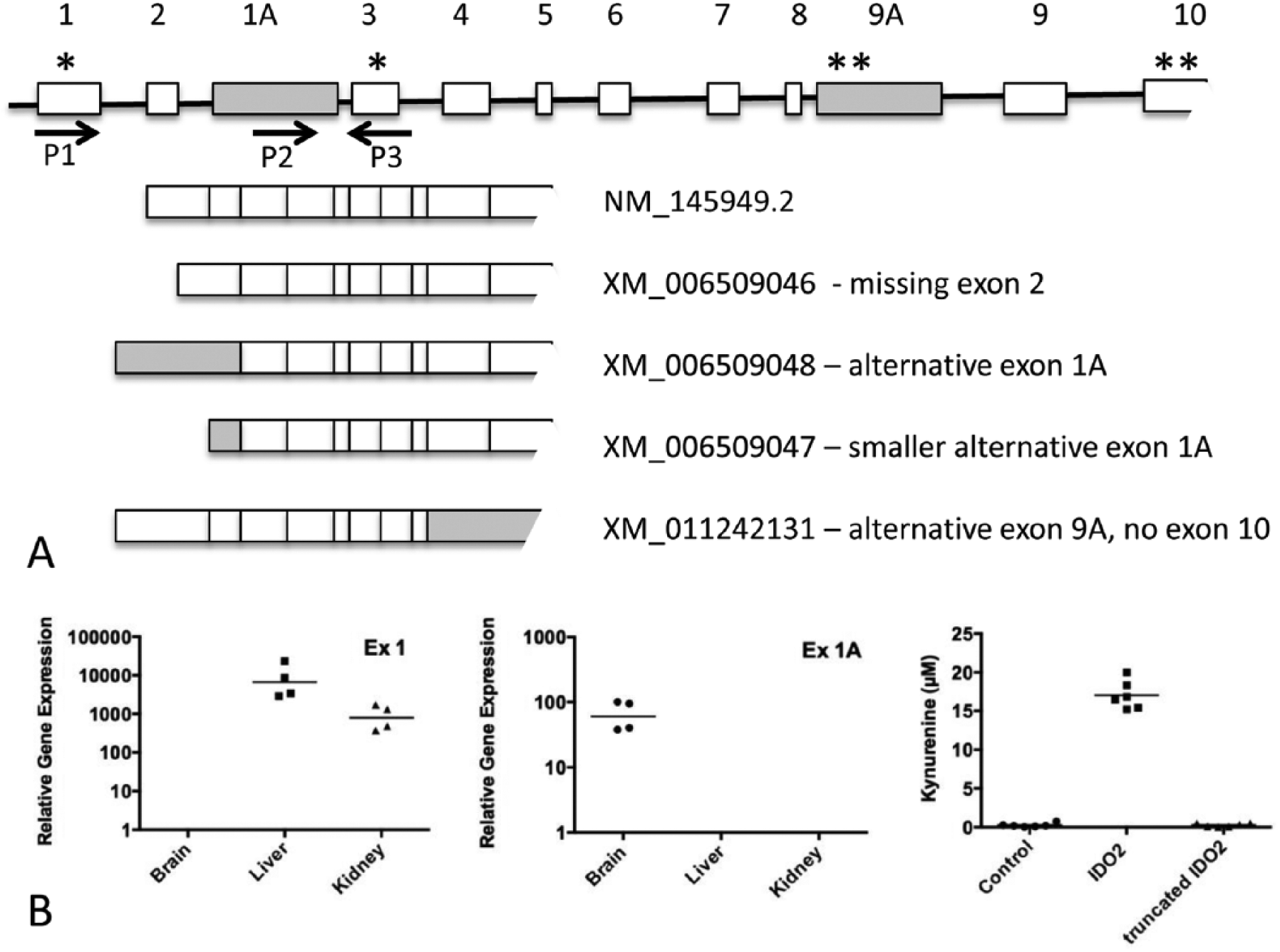
The mouse Ido2 gene generates several alternatively spliced transcripts with distinct tissue distributions and encoding proteins with different enzymatic activities. (A) A schematic figure (not drawn to scale) showing the structure of the Ido2 gene and alternatively spliced transcripts. NM_145949.2 represents the transcript encoding the full-length IDO2 protein. The shaded boxes represent the alternative exons, not found in NM_145949.2. * denotes an actual or potential start codon; ** denotes an actual or potential stop codon. P1, P2, and P3 are primers designed to differentiate between transcripts containing exon 1 (NM_145949.2, XM_006509046, XM_011242131) and exon 1A (XM_006509048). (B) Panels 1 and 2: the transcript containing exon 1 is found in the liver and kidney, whereas the transcript containing exon 1A is found in the brain. Ido2 mRNA was amplified from total RNA extracted from mouse tissues using primers designed to distinguish between exon 1 (P1, P3) and exon 1A (P2, P3). Expression is normalized to the Rpl13a reference gene and the horizontal line represents the geometric mean of expression in 4 mice. Panel 3: HEK293T cells were transfected with expression constructs containing the coding sequence of NM_145949.2 or putative coding sequence of XM_006509048 and enzymatic activity detected by the formation of kynurenine in the cell medium. Two experiments were performed and one is shown with the horizontal bar representing the arithmetic mean of the 6 replicate wells.
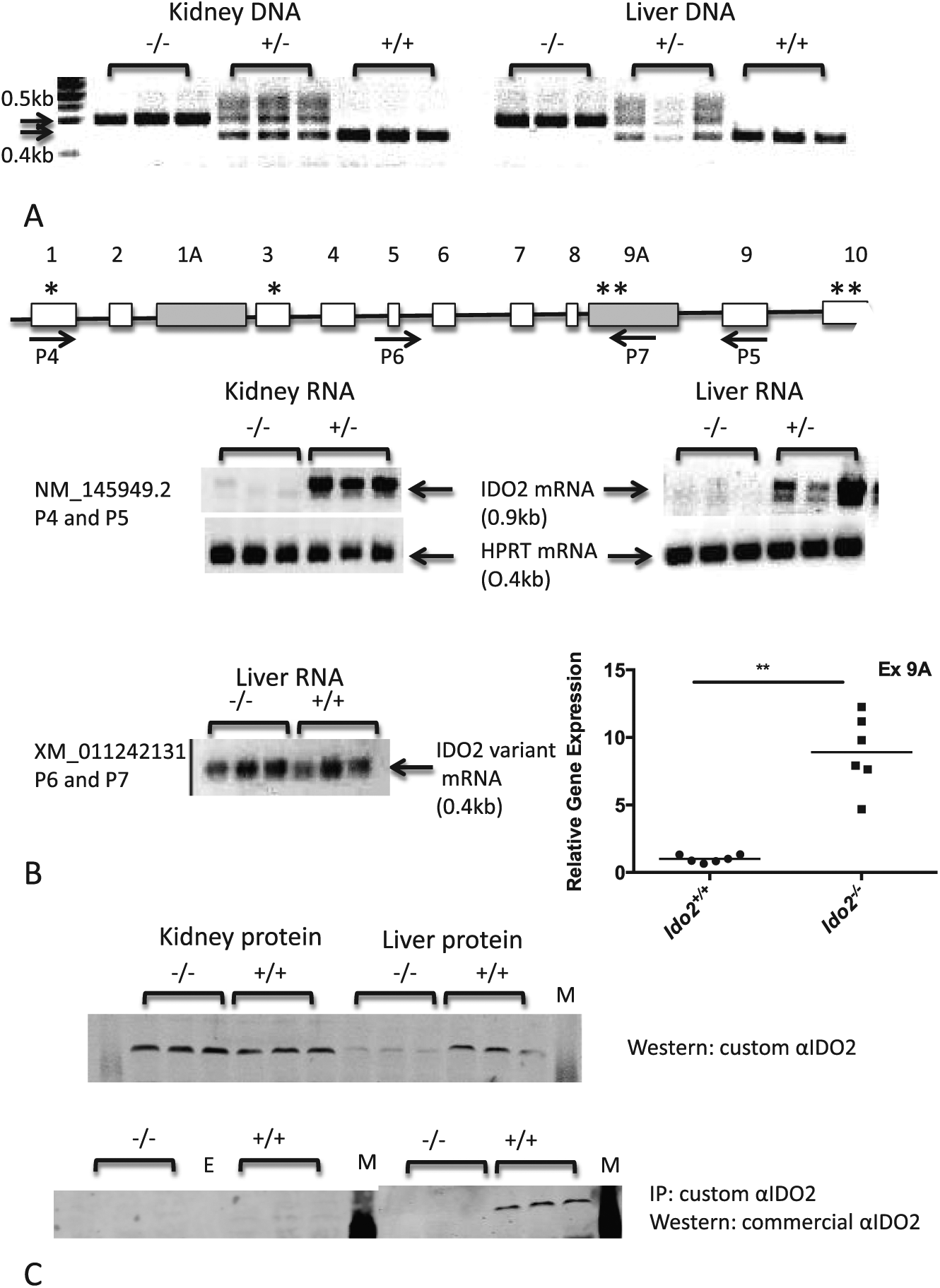
Molecular characterization of Ido2−/− mice. (A) A portion of the Ido2 gene (exon 9/10) is partially deleted in the heterozygous mice and fully deleted in homozygous knockout mice in both kidney and liver tissue. Polymerase chain reaction was performed on genomic DNA using primers distinguishing between the wild-type (0.5 kb product) and knockout (0.4 kb) alleles. (B) Kidney and liver from Ido2−/− mice do not express the full-length Ido2 mRNA. A schematic figure (not drawn to scale) shows the position of the primers used for RT-PCR. The shaded boxes represent the alternative exons, not found in NM_145949.2. * denotes an actual or potential start codon; ** denotes an actual or potential stop codon. Ido2 mRNA, predicted to translate into full-length protein, was amplified using a primer pair (P4 and P5) that has one primer situated within the deleted region of the gene. RT-PCR with a primer pair (P6 and P7), amplifying the alternative transcript, XM_011242131, showed expression in the liver of both wild-type and knockout mice. In addition, RT-qPCR demonstrated a significant upregulation of this transcript in the livers of the knockout mice. Student t test on log-transformed data, **P < .0001, n = 6 mice. (C) Western blot analysis indicates that not all IDO2 antibodies are selective for IDO2. Protein extracted from wild-type and knockout mice was analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis and immunoblotting, with or without immunoprecipitation of the IDO2 protein. Representative individuals (n = 3) are shown from a total of 12 mice/strain. mRNA, messenger RNA; RT-PCR indicates reverse transcription-polymerase chain reaction.
Molecular analysis of Ido2−/− mice
The Ido2-deficient mouse strain used in this study 13 included LoxP sites inserted into the Ido2 gene, with part of the gene deleted by crossing this mouse strain to one expressing a Cre-recombinase transgene. The Cre-recombinase transgene was expressed under the control of the Cytomegalovirus (CMV) promoter, which has activity in most cell types, resulting in common deletion of exon 9/10 of the Ido2 gene. We confirmed the expected patterns of deletion in mouse liver and kidney tissue, with heterozygous mice showing amplification of both alleles and the homozygous mice showing only amplification of either the wild-type or deletion mutant allele (Figure 2A). Reverse transcription-polymerase chain reaction, with primers designed to amplify the sequences within the deleted region, showed that the transcript encoding the full-length Ido2 mRNA is lost in the Ido2−/− mice (Figure 2B). Interestingly, the transcript with the last 2 exons replaced by exon 9A (XM_011242131) should be unaffected by the strategy used to generate the Ido2−/− mice. Confirming this expectation, RT-PCR analysis indicated that this transcript was expressed in the liver of both wild-type and Ido2−/− mice, and RT-qPCR showed that it is significantly upregulated by the gene deletion (Figure 2B, Student t test, P < .0001).
We previously described IDO2 protein expression in mouse tissues using a custom-made anti-IDO2 antibody. 3 This antibody clearly detected a band of approximately 45 kDa that was present in HEK293T cells transfected with an Ido2-expression construct but not in untransfected cells. Western blot analysis of wild-type mouse tissues revealed a band of a similar size in kidney, liver, and reproductive tissues. 3 Verification of these results, using protein from wild-type and Ido2−/− mice, showed that the protein detected with our antibody was diminished in the liver extracts from Ido2−/− mice but was unchanged in the kidney tissue (Figure 2C). This suggests that although our anti-IDO2 antibody does detect the IDO2 protein, it also detects another protein of a similar molecular weight. Although this other protein is not apparent in HEK293T cells and some tissues, for example, brain or heart, it is expressed in the kidney and to a lesser extent in the liver. This protein does not represent a putative protein encoded by XM_011242131 (the Ido2 transcript unaffected by the gene deletion strategy) as that putative protein is only 242 amino acids in length and does not contain the peptide sequence used to generate our anti-IDO2 antibody. The cross-reacting protein was not detected with a custom-made anti-IDO1 antibody (data not shown) and its identity is not known. We tested several mouse anti-IDO2 antibodies and all detected IDO2 protein in transfected cell extracts, but only one antibody (“commercial antibody” or Santa Cruz SC374159; Santa Cruz Biotechnology, in Figure 2C) did not cross-react with a protein of a similar molecular weight to IDO2 in kidney tissue extracts (Figure 2C and data not shown). When IDO2 protein was immunoprecipitated from liver extracts with the custom IDO2 antibody and then detected by the Western blot with the commercial antibody, the band representing IDO2 was completely absent in the Ido2−/− mice (Figure 2C). In comparison, the IDO2 protein was absent or barely detectable in kidney extracts from wild-type mice. Our previous study concluded that the kidney had the highest levels of IDO2 expression 3 but, through verification with the Ido2−/− mice, the current results indicate that the liver has the highest expression of IDO2 protein with there being comparatively little expression in the kidney. The IDO2 protein expression correlates with the higher levels of Ido2 mRNA detected in the liver versus the kidney. 3
The findings described in the first 2 figures have implications for studies examining expression of IDO1 and IDO2. First, RT-qPCR studies using primers downstream of exon 3 of either gene may actually measure the Ido1 and Ido2 mRNA transcripts with alternative first exons, not the transcripts that encode full-length proteins. We could not determine that these alternative transcripts are endogenously translated. Driving translation of the alternative transcripts, by cloning them into an expression vector, resulted in the expression of truncated IDO1 and IDO2 proteins with no detectable enzymatic activity. Therefore, RT-qPCR assays using Ido1 mRNA expression as a predictor of IDO1 enzymatic activity should be designed to have a primer on exon 1. The situation is further complicated in the Ido2 gene with the existence of an additional alternative transcript with a different last exon. Again, it is not known whether the transcript is endogenously translated. If so, it would encode a truncated protein (242 amino acids, truncated at the C terminal), which would be highly unlikely to have enzymatic activity, based on the structure of human IDO1 and its heme-protein interactions. 21 This transcript is still expressed, even at higher levels, in the Ido2−/− mice. It is likely that these mice are null for IDO2 enzymatic activity as the strategy deletes part of the protein required for enzymatic activity.
Ido1 and Ido2 genes are paralogs and the timing of the gene duplication event occurred before the evolution of vertebrates. 6 The genomic structure (position of exons within the coding sequence) has been conserved and it is interesting to note that even the alternative splicing pattern has similarities, with both having transcripts with alternative first exons. It is possible that these alternative transcripts are translated and the putative proteins perform a role unrelated to enzymatic activity. It is also possible that these alternative transcripts directly act on a biological process as is increasingly recognized with long noncoding RNAs. Although long noncoding RNAs are frequently transcribed from intergenic and intronic regions, there are examples of noncoding and coding alternatively spliced transcripts from the one gene having distinct biological roles. 22
Perinuclear and nuclear pattern of IDO2 protein expression in cells
IDO2 protein expression in the mouse previously has been localized to the caput, corpus, and cauda of the epididymis; liver hepatocyte; kidney tubules, Purkinje cells of the cerebellar cortex; and neuronal cells of the cerebral cortex. 23 We previously localized expression of IDO2 protein to kidney tubules and the tails of spermatozoa, using the custom anti-IDO2 antibody described above. 3 We repeated the immunohistochemical localization study using the commercial antibody that was more selective for IDO2 and verified expression with the use of Ido2−/− mice. Although some staining could be seen with the commercial antibody, the pattern was identical in the wild-type and Ido2−/− mice in kidney, brain, colon, testis, and epididymis (SF3). The exception was the liver, where staining was seen in the wild-type mice with anti-IDO2 antibody but not with Ido2−/− mice or isotype control antibody (Figure 3). This study shows a more restricted pattern of IDO2 expression compared with our previous studies and those of others.3,23 Genetically deficient mice are invaluable tools for both assessing the selectivity of antibodies and the specificity of observed staining patterns. Here, it was interesting that the validated positive staining in the liver was mostly perinuclear and nuclear within the hepatocytes. The staining was quantified by counting the number of positive nuclei and measuring the area of staining within each field of vision (Table 1). The subcellular localization of IDO2 protein was investigated in transiently transfected HEK293T cells, but here, the protein expression was cytoplasmic (data not shown).

Perinuclear localization of IDO2 protein in hepatocytes. IDO2 expression was analyzed by immunohistochemistry using a commercial mouse monoclonal antibody. Staining with the monoclonal antibody was observed around the nuclei in (A) Ido2+/+ mice but absent in (B) Ido2−/− mice. No staining was observed in tissue from Ido2+/+ and Ido2−/− mice with the isotype control antibody (C and D, respectively). IDO2 reactivity produced the brown staining from conversion of the diaminobenzidine substrate and the nuclei were stained blue with hematoxylin.
IDO1 protein is not detectable in the liver under normal conditions and elsewhere its expression is cytoplasmic. 24 Surprisingly, despite its abundant expression in liver hepatocytes, little has been reported about the subcellular localization of TDO, although enzyme purification protocols and product information from antibody vendors suggest a cytoplasmic localization. A study examining expression of human IDO1 and TDO noted expression of both enzymes in the cytoplasm of decidualized endometrial stromal cells and epithelial cells in villi. 25 A perinuclear or nuclear subcellular localization, as observed with IDO2 in the liver, has not been reported for IDO1 or TDO and appears to be a distinct feature of IDO2.
IDO2 deletion does not affect Trp metabolism in the liver
Tryptophan metabolism, resulting in the formation of Kyn, is observed when mouse IDO2 is expressed in transfected mammalian cells or recombinant mouse IDO2 protein is used in an in vitro assay.3,8 We confirmed that this was its only detectable effect on amino acid metabolism by assaying the levels of amino acids in the medium of HEK293T cells transfected with mouse IDO2, showing that only Trp was significantly consumed (Figure 4A). However, the expression of IDO2 in the mouse liver had no detectable effect on Trp metabolism, as the tissue levels of Trp and Kyn were the same in wild-type and Ido2−/− mice (Figure 4B). The plasma levels of Trp and Kyn were also similar in wild-type and Ido2−/− mice (Figure 4B). The liver is the major site of IDO2 expression in the mouse, which suggests that IDO2 does not normally contribute significantly to systemic Trp metabolism as reflected in the plasma concentration. In contrast, deletion of the other Trp-metabolizing enzyme results in high levels of circulating Trp. 11 Plasma levels are 10-fold higher in Tdo null mutant mice compared with wild-type mice. IDO2 and TDO showed a similar mRNA expression pattern, with expression becoming detectable in the liver in early neonatal stages and increasing until adulthood (Figure 5A). If IDO2 were important in regulating Trp/Kyn levels in the liver, Ido2 gene deletion might result in a compensatory change in TDO expression. However, the levels of Tdo mRNA were not affected by deletion of the Ido2 gene (Figure 5B, Student t test, P = .95). Ido1 mRNA is undetectable in liver tissue from both strains of mouse. Our results (Figure 4B) and those of Kanai et al 11 suggest that TDO is the major regulator of Trp levels in normal physiology and, despite both proteins being expressed in the liver, IDO2 contributes little, if anything, to Trp metabolism under normal physiological conditions. Another study comparing Kyn and Trp levels in the brains of Ido1−/−, Ido2−/−, and Tdo−/− mice found that Trp levels were elevated only in the Tdo−/− mice, 26 suggesting that TDO is also the major regulator of Trp levels in this tissue.

Exogenous expression of IDO2 increases Trp metabolism; however, deletion of endogenous IDO2 has no significant effect on liver and plasma Trp metabolism. (A) Exogenous IDO2 expression results in Trp depletion. IDO2-trandsfected or control plasmid–transfected HEK293T cells were incubated in medium for 30 hours. After protein precipitation with trichloroacetic acid (TCA), amino acid levels in the medium were measured by HPLC. Of 3 experiments, 1 experiment showing the mean ± SEM of 8 replicate wells is presented (20, 30, and 48 hours, respectively). The only amino acid that was significantly diminished in the medium from the IDO2-expressing cells at any time point was Trp (Student t test performed on the 30-hour time point, **P < .01). (B) Deletion of endogenous IDO2 did not affect Trp metabolism in the liver. Protein was precipitated with TCA from liver tissue homogenates and plasma collected from Ido2+/+ and Ido2−/− mice. Levels of Trp and Kyn were measured in the supernatants by HPLC. No significant differences were observed in Trp or Kyn levels between Ido2+/+ and Ido2−/− mice (Student t test, n = 10 for plasma and n = 12 for liver). HPLC indicates high-performance liquid chromatography.

Tdo and Ido2 have similar mRNA expression patterns; however, although Ido2 deletion changed the expression of a number of genes, it did not affect Tdo mRNA levels. (A) Tdo and Ido2 mRNA expression is low or undetectable at birth and increases in the neonatal period until adulthood. Livers were collected from Ido2+/+ pups at various time points from birth. Expression of Ido2 and Tdo mRNA was measured by RT-qPCR, normalized to the Ywhaz reference gene and expressed as the percentage of expression seen in the adult liver. Data are shown as mean ± SEM, n = 6. (B) Ido2 deletion alters the expression of a number of genes, but Tdo mRNA levels do not change to compensate for the loss of IDO2. The genes (Xdh, Acot1 Cyp4a14, Ehd3, Gdf15, Fam25c Igfbp1, Moxd1) were identified as being differentially expressed in Ido2−/− mice by a microarray analysis on RNA pooled from liver tissues. This was confirmed by RT-qPCR analysis on individual samples using Ywhaz as a reference gene (Student t test on log-transformed data, *P < .05, n = 6). Tdo mRNA was not differentially expressed according to the microarray analysis and this was confirmed using RT-qPCR on individual samples. Data are expressed as the mean of the log-transformed ratio of expression in Ido2−/− liver relative to Ido2+/+ liver ± SEM. mRNA indicates messenger RNA; RT-qPCR, quantitative reverse transcription-polymerase chain reaction.
Analysis of Ido2 gene deletion phenotype
Analysis of the normal phenotype of Ido2−/− mice revealed no obvious differences to Ido2-+/+ mice consistent with other studies of this strain that suggest functional roles only in chronically stressed or disease states.13,18,19 As the Trp-metabolizing enzymes play a role in regulating maternal tolerance responses during pregnancy, 27 we examined the expression of the 3 enzymes in the placenta and yolk sac. Ido2 mRNA was barely detectable in the placenta, although some expression was observed in the yolk sac (SF4A). In comparison, Ido1 and Tdo mRNAs were more highly expressed in both tissues. Ido2 mRNA has been reported in the mouse uterus early in pregnancy, and overexpression resulted in downregulation of decidualization marker genes, suggesting that it may play a role in implantation. 28 However, the breeding of Ido2−/− and Ido2+/+ mice appeared similar, with no noticeable differences/problems with litter frequency or pregnancy loss. Litter sizes and sex ratios were similar in Ido2−/− and Ido2+/+ mice (SF4B), as were the weights of the mice (SF5A). Histopathologic examination of tissues, including liver, revealed no morphological differences (SF5B), and no statistically significant differences were observed in plasma analysis using a comprehensive metabolic panel (SF6).
Global gene expression in the liver was investigated by array analysis using liver RNA samples pooled from either Ido2+/+ or Ido2−/− mice. This was an exploratory analysis using pooled samples, so genes were selected for verification by RT-qPCR on individual samples, based on availability of functional information and degree of differential expression as determined by the array analysis. The selected genes confirmed to be differentially expressed with a Student t test are shown in Figure 5B. The small number of differentially expressed genes identified does not suggest the involvement of IDO2 in a specific pathway. Xanthine dehydrogenase (XDH; P < .001), one of the interconvertible forms of xanthine oxidoreductase, is a nicotinamide adenine dinucleotide (NAD)-dependent cytosolic dehydrogenase, capable of metabolizing hypoxanthine to xanthine. 29 A common link between the Kyn pathway and XDH is that the former supplies NAD+, whereas the latter requires NAD+ for activation. As Ido2 deletion does not affect Trp/Kyn levels in the liver, it might be expected to also have minimal effects on downstream metabolites, such as NAD+, suggesting that a possible regulation mechanism through this pathway is unlikely. Other regulators of Xdh expression include inflammatory stimuli such as cytokines and lipopolysaccharide.30,31 Gdf15, also found upregulated in Ido2−/− mouse liver lysates relative to Ido2+/+ (P = .021), is the murine orthologue of the human macrophage inhibitory cytokine-1 (MIC-1), discovered as a divergent member of the transforming growth factor β superfamily. Gdf15 is upregulated in hepatocytes in response to injury or stress. 32 Cyp4a14 (P = .018) and Acot1 (P = .006), both upregulated in the absence of IDO2, are, respectively, involved in fatty acid degradation and biosynthesis of unsaturated fatty acid. These genes also are upregulated in the liver of a mouse model of stress in a mechanism dependent on the peroxisome proliferator–activated receptor α signaling pathway. 33
Downregulation of Moxd1 (P = .011) and upregulation of Igfbp1 (P = .049), as observed in the Ido2−/− mice, occurs in cases of reduced insulin bioavailability.34,35 Insulinlike growth factor–binding proteins, such as the differentially expressed IGFBP1, sequester insulinlike growth factors which in turn maintain glucose homeostasis by inhibiting glucose-stimulated insulin secretion at higher concentrations and stimulating insulin release at lower concentrations. Interestingly, IDO2 overexpression in uterine stromal cells was found to downregulate Igfbp1 mRNA, which is a marker for decidualization in this system. 28 Moxd1 is one of the most downregulated genes in the livers of BALB/c mice fed a high-fat diet. 36 Ehd3, upregulated in Ido2−/− mice (P = .001), has been implicated in endocytic recycling and tubular structures, directing their motility. The least understood protein in the list is Fam25c (P = .001), which is a predicted sequence with no reports on its structure or role thus far.
Although no conclusions can be drawn about the pathways regulated/affected by IDO2 deletion, it is possible that stress/inflammatory responses are leading to the induction of genes such as Gdf15 and Xdh. In addition, the alterations in other genes (Acot1, Cyp4A14, MoxD1, Igfbp1) may suggest altered lipid/glucose metabolism. No difference in weights or liver pathology were observed up to early adulthood, but it would be of interest to investigate obesity, glucose metabolism, and inflammation as Ido2−/− mice age.
Does IDO2 possess abilities unrelated to its enzymatic activity?
Despite IDO2 having enzymatic activity in in vitro assays and when exogenously expressed in mammalian cells, it does not have a detectable effect on Trp/Kyn metabolism in vivo in normal physiology. This is in contrast to the other Trp-catabolizing enzyme expressed in the liver, TDO, whose deletion has a dramatic effect on circulating Trp levels. IDO2 is also expressed in the liver but it cannot compensate for the deletion of Tdo in maintaining normal Trp homeostasis. In comparison, Ido1 deletion has modest effects on Trp/Kyn in the plasma 26 and the enzyme is not expressed in the liver in normal physiology. Through induction by inflammatory stimuli, IDO1 has significant effects on the Trp/Kyn balance, both systemically and in the microenvironment. 37 IDO2 may also make a significant contribution to Trp metabolism only in certain, as yet, undiscovered conditions. 9 Similar to IDO1, this may be related to induction in different cell/tissue types by certain stimuli. One observation of this study was that transient overexpression in a cell line produced both cytoplasmic IDO2 expression and detectable enzymatic activity, whereas the nuclear-associated staining of IDO2 in the liver of Ido2+/+ mice did not correlate with any difference in Trp/Kyn levels. It is possible that the subcellular localization of IDO2 regulates its enzymatic activity by affecting its interactions with cofactors. For example, cytochrome b5 is a cofactor for the activity of IDO2 in vitro 8 and its expression in the liver is mostly in the membrane-bound forms found in the endoplasmic reticulum and outer mitochondrial membranes. 38
Another possibility is that Ido2 has a role unrelated to enzymatic activity. Heme oxygenase-1 (HO1) has biological activity in the nucleus unrelated to the metabolism of heme to generate biliverdin and carbon monoxide. C-terminal cleavage of the enzyme results in an enzymatically inactive protein that translocates to the nucleus, where it interacts and regulates several transcription factors and DNA repair enzymes. 39 Although IDO2 does not have the particular signaling capability possessed by IDO1, 7 regulation of protein function by interaction might conceivably be a function of IDO2 and warrants further investigation.
Although IDO2 shares with TDO the characteristic of constitutive expression in the liver, in many other ways, it is more similar to IDO1. These features include the following: an alternative splicing pattern potentially generating truncated proteins, induction by inflammatory stimuli in certain cell types, involvement in some inflammatory processes, and having no/modest effects on physiological Trp homeostasis. IDO1 differs from IDO2 in that its enzymatic activity has been demonstrated to mediate some of its biological effects, through the use of pharmacological inhibitors and Trp supplementation. IDO2-selective inhibitors have been identified, 40 but other studies have mostly used genetic approaches to modulate IDO2 that would affect both enzymatic and potential nonenzymatic activities.13,41 The low enzymatic efficiency of IDO2 in in vitro/cellular assays and the unchanged Trp/Kyn levels in the liver in Ido2−/− mice suggest that IDO2 is not as efficient as IDO1 and TDO in converting Trp to Kyn in vivo. It remains possible that the enzyme may contribute to Trp metabolism in specific circumstances or locations. Alternatively, IDO2 may possess a different enzymatic activity or signaling function.
Conclusions
The murine Ido2 gene generates alternatively spliced Ido2 mRNA transcripts. We identified an alternate first exon in murine Ido2, the use of which generates mRNA transcripts that are predicted to generate IDO2 proteins which lack an enzymatic function. In mice, IDO2 protein is strongly expressed only in hepatocytes, with a perinuclear and nuclear distribution. Deletion of the Ido2 gene does not have a major impact on normal reproduction or mouse phenotype, up to the young adult stage at least, unless chronic stress or disease is present.13,18,19 Plasma, liver, and brain concentrations of Kyn and Trp are unchanged in Ido2−/− mice under normal physiological conditions, consistent with the concept of a role in stress or disease responses. Although IDO2 plays a role in some pathological conditions,13,18,19 a physiological role, if any, remains uncertain.
Footnotes
Acknowledgements
The authors wish to acknowledge the assistance of the Bosch Advanced Microscopy Facility and Molecular Biology Facility, as well as the Histopathology Laboratory of the Discipline of Pathology. They are also grateful for the assistance of Ghassan Magzhal, Cacang Suarna, Roland Stocker, and Brett Garner in measuring amino acids and metabolites.
Peer review:
Four peer reviewers contributed to the peer review report. Reviewers’ reports totaled 745 words, excluding any confidential comments to the academic editor.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Australian Research Council.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
HJB, NHH, FFJ, SMB, SW, STF, and LKT conceived and designed the experiments and analyzed the data. HJB wrote the first draft of the manuscript. NHH and FFJ contributed to the writing of the manuscript. GCP and RM provided advice and reagents. All authors reviewed and approved the final manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.