
Abstract
Cervical cancer remains a major global health burden, with high-risk human papillomavirus (HPV), particularly HPV-16, responsible for most cases. While licensed prophylactic HPV vaccines are effective, their type-restricted coverage and lack of therapeutic activity highlight the need for next-generation vaccine strategies. In this study, a systems vaccinology-based approach was used to design a multi-epitope vaccine with both prophylactic and therapeutic potential against HPV-16. Immunodominant epitopes eliciting immune responses from HPV-16 E5, E6, E7, and L2 proteins were identified using immunoinformatics methods. Transcriptomic and pathway enrichment analysis of cervical cancer data sets identified interleukin-17A (IL-17A) signaling as a favorable immune axis, supporting its inclusion as a cytokine adjuvant. Population coverage evaluation demonstrated broad global applicability, with a predicted coverage of 99.98% across MHC class I and II alleles. Among 7 designed constructs, 1 candidate (model D) exhibited favorable physicochemical and structural properties, including high predicted solubility (73%), strong model quality, and antigenic potential. Docking analyses indicated strong correlations between native complexes and the corresponding vaccine-IL-17 receptor A and C complexes (Pearson r = .98 and .88, respectively). Molecular dynamics simulations over 500 ns confirmed structural stability, with favorable MM/PBSA binding free energies (ΔG_total ≈ −31 kJ/mol). Immune response simulations demonstrated activation of CD4+ and CD8+ T-cells, memory B-cell formation, and a predominantly IFN-γ immune response. These findings demonstrate that the proposed construct is a promising HPV-16 vaccine candidate; nevertheless, targeted in vitro and in vivo studies are essential to confirm its immunogenicity, efficacy, and safety prior to further clinical developments.
Introduction
Cancer, one of the most critical global healthcare issues, causes an extensive number of deaths annually and substantially lowers the quality of the patient’s life. Cervical cancer is the fourth most common malignancy in females worldwide. 1
Multiple risk factors are associated with the incidence of cervical cancer. However, the leading cause of more than 90% of the cases is human papillomaviruses (HPVs). 2 Nearly 70% of cervical cancers result from high-risk subtypes of HPVs (HR-HPVs), including HPV-16 and HPV-18, while some other subtypes might cause genital warts. 3
Nowadays, available preventive vaccines have helped in reducing the incidence of HPV-caused cervical cancers. Moreover, advancements in knowledge, screening techniques, and standard preventive methods (such as Pap smear and liquid-based cytology) facilitated the detection of pre-cancerous lesions and early-stage malignancy, leading to a decrease in invasive cervical cancer cases. 4 Nevertheless, the burden of cervical cancer remains troubling, especially in low-income and middle-income countries. 1 The development of effective prophylactic and therapeutic HPV vaccines is currently a critical, reliable solution. 5
The HPV genome encodes various functional regions, including early (E) and late (L) proteins. E proteins, E1-E7, are vital in the replication and pathogenicity of the virus, while L1 and L2 proteins are essential capsid proteins, and L1 contributes to the vaccine assembly and encapsulation.4,6
Present prophylactic HPV vaccines, developed via recombinant DNA technology, use L1 capsid proteins to form virus-like particles (VLPs). Cervarix is bivalent (HPV-16 and 18), Gardasil is quadrivalent (effective against HPV-16, 18, 6, and 11), and Gardasil-9 is nine-valent, protecting against 5 additional strains (HPV-31, 33, 45, 52, and 58). 3 Despite the worldwide commercial availability of these vaccines, the demand for HPV vaccines, particularly in low- and middle-income countries, is still critical. 4
While these vaccines have shown high efficacy in preventing HPV infections, they possess several limitations, including type-restricted protection, lack of therapeutic capabilities, cold-chain dependence, limited accessibility in developing countries, and inability to induce broad-spectrum immunity across different HPV subtypes. 7
Although the immunogenicity of the L1 capsid protein is favorable (compared with the shorter L2 capsid protein), it is less conserved across HPV types. Therefore, using L2 capsid protein in vaccines could induce immunologic responses with a wider coverage range. However, applying L2 protein -as peptide segments alone- leads to limited immunogenicity. To enhance the generated immune responses, adjuvants are needed.2,6 Moreover, all the commercially available HPV vaccines only confer prophylactic effects, as they contain only the L1 capsid protein. Developing therapeutic HPV vaccines could be a critical step in decreasing cervical cancer burden. Thus, E proteins (critical HPV pathogenic proteins) have been used in recent research to develop therapeutic vaccines.4,5 Despite some investigations, no therapeutic vaccine has been approved yet.
Recently, the reverse vaccinology (ie, employing protective epitope sequences) approach has emerged as an efficient and economically advantageous strategy for vaccine design. Polypeptide vaccines, which are made of a string of epitope subunits, are inherently less immunogenic, so using an appropriate adjuvant is necessary. Adjuvants induce macrophages, neutrophils, and dendritic cells (DCs) of the innate immune system, which further results in stimulating adaptive immunity. 8
Immunoinformatics-based tools offer valuable perspectives on epitope identification, vaccine efficacy and stability, adjuvant selection, antigenicity, and allergenicity and are frequently used to design polypeptide vaccines against various pathogens. 5
In contrast with prior prophylactic HPV vaccines whose designs are focused on L1 capsid proteins or limited HPV-E antigens, this study integrates E5, E6, E7, and L2 to target both early and late stages of HPV-16 infection, thereby supporting both therapeutic and prophylactic vaccine design objectives. Furthermore, computational HPV vaccine design studies rely primarily on reverse vaccinology and often lack integration of immune network context. To address this limitation, we apply a systems vaccinology-guided computational framework that introduces potential adjuvants, integrates viral epitope selection via reverse vaccinology, and incorporates structural modeling and simulation to design a multi-epitope vaccine against HPV-16.
Materials and Methods
The methodology used to design the vaccine structure was directed in the following steps.
Extraction of sequences and their primitive physiochemical attributes
The sequences of E5, E6, and E7 oncoproteins of HPV-16, as well as HPV16-L2 capsid protein, human IL-17A, and universal TpD polypeptide, were retrieved from the UniProt database (https://www.uniprot.org/). 9 The physiochemical features of each sequence were analyzed via the ProtParam tool of Expasy (https://web.expasy.org/protparam/). 10 The obtained data are presented in Supplementary Table S1.
Phylogenic analysis among HPV-16 selected proteins and high-risk HPV species
To discover the extent of similarity between the selected sequences of HPV-16 (Taxid: 333760) and 11 HR-HPVs, the NCBI BLAST was used (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch). Using standard databases, nucleotide collection nr/nt was implemented for the search setup, and the reward/penalty ratio for match-mismatch of the sequences (that are at least 75% conserved) was set to be 1/-1. Subsequently, MEGA (v.X) was used to align all the sequences using the ClustalW method. Phylogenic analysis was carried out using the neighbor-joining approach with a bootstrap of 1000. 11
HLA allele retrieval
To increase the specificity of the immune response, frequent HLA alleles (worldwide) were selected using the allele frequency net database (http://www.allelefrequencies.net) and a meta-analysis covering 497 population studies.12,13 Furthermore, alleles that are considered risk factors for HPV-16-associated squamous cell cervical cancer were collected from another meta-analysis conducted by Madeleine et al 14 (data available in Supplementary Table S2).
Predicting epitopes with affinity toward MHC-1 and MHC-2
Three servers were employed to identify the potential MHC-1-binding epitopes including (1) EpiJen (http://www.ddg-pharmfac.net/epijen/EpiJen/EpiJen.htm; selecting peptides within the top 5% scores) which predicts MHC-I binding epitopes using a multi-step algorithm combining proteasomal cleavage, TAP transport, and MHC binding efficiency, (2) IEDB (http://tools.iedb.org/mhci/; selecting peptides ranked ⩽1% as classified strong binders) which identifies MHC-I binders using consensus prediction methods integrating multiple algorithms, and (3) NetMHC 4.0 (http://www.cbs.dtu.dk/services/NetMHC/; considering peptides with percentile ranks ⩽ 0.5% as strong binders) which predicts MHC-I binding affinity using artificial neural networks.15-17 The average length of the harvested sequences was 8-10 amino acids across the employed tools.
To predict epitopes with affinity toward MHC-2, 3 online tools were used including (1) NetMHCIIpan 4.0 (http://www.cbs.dtu.dk/services/NetMHCIIpan/; selecting peptides with percentile ranks ⩽ 2%) which predicts MHC-II binding epitopes using pan-specific neural network model, (2) IEDB (http://tools.iedb.org/mhcii/; retaining epitopes with percentile ranks ⩽ 2% as strong binders) which applies consensus prediction approaches for MHC-II binding, and (3) TepiTool (http://tools.iedb.org/tepitool/; selecting peptides ranked ⩽ 10% as potential binders) which identifies promiscuous MHC-II epitopes using IEDB-based prediction pipelines.16,18,19 The average length of the predicted epitopes was in the range of 12-25 amino acids across the employed tools.
Predicting epitopes with affinity toward cytotoxic T-cells and B-cells
In this step 4 servers were employed to detect the CTL-binding antigenic regions including (1) CTLpred (http://crdd.osdd.net/raghava/ctlpred/about.html; default settings to select 9-mer epitopes was applied) which predicts epitopes using machine-learning models based on amino-acid composition and motif information to classify peptides as CTL binders or nonbinders, (2) PAComplex (http://pacomplex.life.nctu.edu.tw/) which identifies epitopes by modeling peptide-MHC-TCR interaction compatibility, prioritizing peptides with favorable structural and physicochemical interaction scores, (3) NetCTLpan 1.1 (http://www.cbs.dtu.dk/services/NetCTLpan; using default thresholds for selecting 8-14-mer peptides) which predicts CTL epitopes using a pan-specific neural network that integrates MHC-I binding, proteasomal cleavage, and TAP transport efficiency, and (4) HPVdb (http://cvc.dfci.harvard.edu/hpv/HTML/viewEpitope.php) which provides experimentally validated and predicted HPV-specific CTL epitopes curated from literature and immunological databases.20-23
Subsequently, 3 servers including (1) BepiPred 3.0 (https://services.healthtech.dtu.dk/services/BepiPred-3.0/; epitopes selected based on the highest epitope score percentage) which predicts linear B-cell epitopes using a deep-learning model trained on experimentally validated antibody-antigen structures, (2) ABCpred (https://webs.iiitd.edu.in/raghava/abcpred/ABC_submission.html; default threshold of 0.51 and epitope length of 16 amino acids was applied) which identifies linear B-cell epitopes using an artificial neural network, and (3) LBtope (https://webs.iiitd.edu.in/raghava/lbtope/protein.php; selecting peptides of 5 to 30 amino acids with a minimum prediction probability of 60%) which predicts linear sequences using a support vector machine (SVM) trained on validated B-cell epitope data sets, were employed to discover B-cell linear epitopes.24-26
Population coverage analysis of obtained epitopes
Population coverage analysis was performed using the Immune Epitope Database (IEDB) population coverage tool (https://tools.iedb.org/population/) which estimates population coverage based on an allele frequency-weighted probabilistic model. 27 Each selected epitope was evaluated individually against the predefined HLA allele panel, which included both MHC class I and class II alleles. For each epitope, the projected population coverage, average number of epitope-HLA combinations recognized per individual (average hit), and PC90 (minimum number of epitope-HLA combinations recognized by 90% of the population) were calculated for the global population. The results were summarized in supplementary Figure S2.
Selecting epitope segments
The harvested sequences of the steps mentioned above were collected, and immunodominant regions with significant overlap were pooled to construct a vaccine structure.
Predicting toxicity, allergenicity, and antigenicity of pooled epitopes
Toxicity of the vaccine construct was assessed using ToxinPred tool (https://webs.iiitd.edu.in/raghava/toxinpred/multi_submit.php), which predicts peptide toxicity using an SVM model trained on Swiss-Prot data sets, applying the default SVM threshold of 1.0 to classify sequences as toxic or nontoxic. 28 Allergenicity of the vaccine construct was evaluated using AllerCatPro v.2.0 (https://allercatpro.bii.a-star.edu.sg/), which predicts allergenic potential by comparing sequence and structural similarity to known allergens based on FAO/WHO guidelines, using the default parameters. 29 The antigenicity of the sequences was studied using VaxiJen v.2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) which evaluates antigenicity through an alignment-independent physicochemical model, considering sequences with scores ⩾0.5 as antigenic. 30 The obtained data are presented in Table 1.
Pooled antigen sequences of HPV-16 along with allergenicity, antigenicity, and toxicity attributes; accordingly, all listed epitopes were predicted to be nonallergenic and nontoxic while exhibiting favorable antigenicity scores, supporting their suitability for inclusion in the multi-epitope vaccine construct.

Identification of a proper adjuvant via systems vaccinology approaches
A microarray-based gene expression profile was used to identify critical genes (whose expression profile changed the most under cervical malignant circumstances) and conduct molecular network modeling. The expression data retrieved from the den Boon et al 31 study (ID: GSE63514), was based on GPL570 Affymetrix human genome U133 plus 2.0 array platform and downloaded from the gene expression omnibus (GEO) database of NCBI (https://www.ncbi.nlm.nih.gov/geo/). 32
The harvested samples were normalized, and correspondent differentially expressed genes (DEGs) were identified using RStudio (v.06) by implementing “affy” (v.1.7), “simpleaffy” (v.2.64), “MASS” (v.7.3), and “limma” (v.3.48; Benjamini-Hochberg false discovery rate method and adjusted P-value < .05 were applied) R packages.
Differentially expressed genes with more than 100 folds of upregulation were used as the inputs of the Enricher tool (https://maayanlab.cloud/Enrichr/; which maps DEGs with >100-fold upregulation to curated pathway databases, using enrichment significance based on adjusted P-values) 33 and the database of the Kyoto Encyclopedia of Genes (KEGG; https://www.genome.jp/kegg/) 34 was employed to conduct pathway enrichment. The STRING server (https://string-db.org/) 35 was used to extract all predicted associations among proteins and generate a PPI network. To explore the subnetwork and interacting receptors within the IL-17A signaling pathway (one of the most enriched pathways; P-value = 2.35e−4), CytoHubba (https://apps.cytoscape.org/apps/cytohubba), a Cytoscape (v.3.8) plugin which uses topological ranking algorithms and focuses on the most significantly enriched pathways, was implemented. 36
The Kaplan-Meier plot was used to analyze the overall survival of patients with low and high extent of IL-17A expression (who were diagnosed with either squamous cell carcinoma of the cervix, endocervical adenocarcinoma, or uterine carcinosarcoma). For this purpose, the GEPIA2 online tool (http://gepia2.cancer-pku.cn/) was implemented, which uses the Log-rank test (or Mantel-Cox) to calculate survival distributions. 37
Designing the constructs and evaluation of their properties
By joining selected epitopes, TpD sequence, linkers (EAAAK-oligomers), and human IL-17A (chains A and B), a total of 7 vaccine structures were proposed (data available in Supplementary Table S3). The next step was to assess the designated vaccines’ physiochemical properties, solubility, allergenicity, and antigenicity.
ProtParam of Expasy (https://web.expasy.org/protparam/) was applied to calculate the GRAVY index (or grand average of hydrophobicity; an indicator of hydrophilicity), aliphatic index (an indicator of thermostability), theoretical pI, and molecular weight of the sequences. To assess the solubility of the vaccines, the SOLpro (http://scratch.proteomics.ics.uci.edu/; prediction score ⩾ 0.5) tool was employed, which predicts protein solubility upon overexpression using a sequence-based machine-learning model and anticipates the solubility with 74% accuracy. 38 Subsequently, 2 online tools, (1) AllerTOP v.2.0 (http://www.ddg-pharmfac.net/AllerTOP/) which predicts allergenicity using an alignment-free, physicochemical property-based machine-learning model to classify peptides as allergenic or nonallergenic 39 and (2) AlgPred (https://webs.iiitd.edu.in/raghava/algpred/) which combines SVM-based classification, amino-acid composition, and similarity to known allergen motifs using predefined decision thresholds, 40 were used to evaluate the allergenicity of the epitopes. The antigenicity of the sequences was studied using 2 servers, including ANTIGENpro (http://scratch.proteomics.ics.uci.edu/) which predicts antigenicity using a sequence-based machine-learning model, with scores ⩾0.5 indicating antigenic potential 41 and VaxiJen v.2.0 30 (with the corresponding parameters and cutoff values described in Section 2.8).
Homology modeling
The 3D modeling of the sequences was conducted via GalaxyTBM (https://galaxy.seoklab.org/cgi-bin/submit.cgi?type=TBM) which performs template-based protein structure modeling by identifying homologous structures from the Protein Data Bank and refining the target sequence using default alignment and template selection. 42 The modeled structures were then subjected to GalaxyRefine (https://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE) modification, which is based on measuring global distance test (GDT) and root mean square deviation (RMSD) indexes.
To exert the validation of the refined models, 3 online tools were used: (1) ProSA-web (https://prosa.services.came.sbg.ac.at/prosa.php) which validates model quality by calculating a knowledge-based Z-score that reflects overall structural plausibility compared with experimentally determined protein structures, 43 (2) PROCHECK (https://www.ebi.ac.uk/thornton-srv/software/PROCHECK/) which assesses stereochemical quality using Ramachandran plot analysis, evaluating residue distributions under standard validation criteria, 44 and (3) ERRAT (https://saves.mbi.ucla.edu/) which evaluates nonbonded atomic interactions using a statistical error function, reporting an overall quality factor based on default thresholds. 45
Prediction of conformational epitopes of B-cells
Discontinuous B-cell epitopes were identified on the refined 3D models of vaccine candidates by the ElliPro server (http://tools.org/ellipro/; he default maximum distance of 6 Å was applied) which predicts epitopes based on protein 3D structure by approximating the protein shape as an ellipsoid and calculating residue protrusion indices. 46
Immune response simulations
In silico immune response simulations were performed to evaluate the immunogenicity of the designed multi-epitope vaccine using C-ImmSim tool (https://kraken.iac.rm.cnr.it/C-IMMSIM/index.php?page=1) which simulates mammalian adaptive immune responses based on position-specific scoring matrices and immune system dynamics. 47 A 3-dose vaccination regimen was simulated using the default biological calibration of the platform (8 hours per time step). Accordingly, antibody responses, B- and T-cell population dynamics, cytokine profiles, and immune memory formation were analyzed to assess vaccine-induced humoral and cellular immunity.
Cross-docking between vaccine candidates and IL-17 receptors A and C (IL-17RA and IL-17RC)
To identify the most eligible construct, docking simulations were carried out among 7 vaccine constructs and 2 receptors (IL-17RA and IL-17RC, which were extracted from the harvested IL-17 A signaling network). Moreover, the isolated native IL-17A structure was docked with both IL-17RA and IL-17RC to be considered as a reference to study correlations among the resulting docking energies. HADDOCK v.2.4 (https://wenmr.science.uu.nl/haddock2.4/) flexible docking method was performed, which includes a variety of predictive and empirical information (eg, NMR studies and residual dipolar coupling). 48 Potent ligand-receptor interactions established in the binding pocket of model D-IL17RA/RC complexes were further explored using the LigPlot+ software (Supplementary Table S5). 49
Molecular dynamics simulation and binding free energy analysis
Molecular dynamics simulations were carried out to gain better insight into the protein-protein interaction at the molecular level. Two complexes, which constituted structure D (the most correlated structure to the reference model, as seen in the cross-docking analysis) and IL-17RA or IL-17RC, were subjected to 500 ns of MD simulation analysis. GROMACS 5.1.2 was used to exert the simulation via the AMBER99SB forcefield. Each protein complex was solved in TIP3 P water and the physiological concentration of NaCl. 50 Methods Verlet and PME were implemented to integrate the particles’ motion calculations and electrostatic equations, respectively. The energy minimization (EM) step was directed under the steepest descent algorithm. Two steps of NVT (LINCS algorithm was applied to impose H-bond optimization) and NPT ensembles were implemented, and the final MD run was performed for 500 ns. 50
Binding free energies were computed using a GROMACS-compatible MM/PBSA workflow, in which the total binding free energy (ΔG_bind or ΔG_total) was decomposed into van der Waals (ΔE_vdW), electrostatic (ΔE_elec), polar solvation (ΔG_polar), and nonpolar solvation (ΔG_nonpolar) components. To this end and for both complexes, MM/PBSA calculations were performed on 800 snap shots, extracted every 500 ps, from the last 400 ns of the equilibrated MD trajectories. The polar solvation energy was calculated using the Poisson-Boltzmann (PB) model, while the nonpolar solvation contribution was estimated from solvent-accessible surface area (SASA).
Codon optimization and in silico cloning
Structure D was uploaded to the EMBOSS backtranseq (https://www.ebi.ac.uk/Tools/st/emboss_backtranseq/) to harvest the corresponding cDNA. The next step was to increase the adaptability of the sequence to E. coli K12 host. Therefore, the JCat tool (https://www.jcat.de/) was used, and the codon adaptation index (CAI) was optimized to imply higher protein expression. 51 Plasmid pQE-30, which is resistant to chloramphenicol and ampicillin, was the selected vector. Thus, the restriction sites of BamHI (5’-GGATCC-3’) and HindIII (5’-AAGCTT-3’) were joined by the N-terminal and C-terminal of the vaccine sequence, respectively. To ensure that the enzymes mentioned above are zero-cutters, the designated sequence was evaluated by the NEBcutter v.3.0 tool (https://nc3.neb.com/NEBcutter/).
Results
Extracting sequences and their primitive physicochemical attributes
The sequences of HPV-16 post-translational (E5, E6, E7) and small capsid proteins (L2) and both chains of IL-17A (extracted from UniProt) together with corresponding physicochemical values (calculated using ProtParam) are presented in the Supplementary Table S1.
Phylogenic assessments of the selected HPV epitopes
To compare selected protein sequences of HPV-16 with other HR-HPV types, the L2, E5, E6, and E7 sequences were analyzed using a basic local alignment search (BLAST) for each sequence by the NCBI database. No significant similarity between the E5 protein of HPV-16 and the proteins from other HPV types were found. The phylogenic assessments of the proteins were conducted by MEGA-X software, and the pairwise-distance score was calculated (based on Maximum likelihood, lower extents indicate more phylogenic similarities). Resultantly, HPV-16 E6 was mainly similar to the E6 of HPV-35, -33, -58, -31, and -73. The most similar proteins to HPV-16 E7 and L2 were found in HPV-31 and -35 and HPV-35, -31, -33, and -58, respectively. The corresponding phylogenic trees were illustrated using the neighbor-joining method and based on pairwise distance indexes (Figure 1).
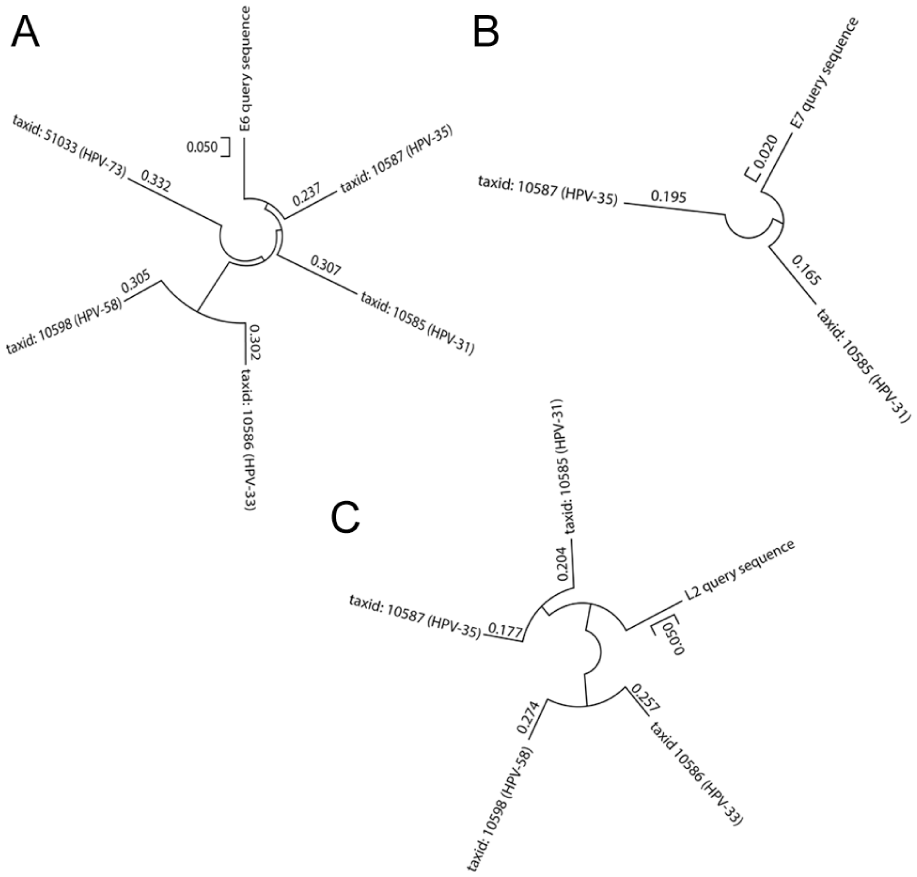
Phylogenetic similarity assessment of HPV-16 proteins to other high-risk HPV types; the results of HPV-16 (A) E5, (B) E7, and (C) L2 proteins show their closest similarities to corresponding proteins from other HPVs, with lower pairwise distances indicating greater evolutionary relatedness.
Predicting epitope sequences with mutual affinity toward MHC-I, MHC-II, cytotoxic T-cells, and B-cells
Selecting MHC-I binding antigens was directed using 3 different online tools to ensure higher accuracy. Similarly, to detect MHC-II, CTL, and linear B-cell epitopes, 3, 4, and 3 predicting servers were employed, respectively. Common immunodominant antigens among all harvested sequences were retrieved to construct vaccine sequences (Table 1). All predicted epitopes from HPV-16 E5, E6, E7, and L2 proteins were classified as nonallergenic and antigenicity analysis showed that all sequences exceeded the threshold for antigenicity (with scores ranging from 0.41 to 1.66). Toxicity screening using the ToxinPred SVM (Swiss-Prot) model predicted all epitopes as nontoxic, with SVM scores below the toxicity threshold (Table 1).
Assessment of HLA allele coverage and population applicability of selected epitopes
The specificity of the immune response was significantly increased by selecting frequent HLA alleles using the allele frequency net database (which identified 35 HLAs) and 497-population-covering meta-analysis data (which identified 19 HLAs).12,13 The selected HLA alleles were cross-referenced with those identified as risk factors for HPV-16-associated squamous cell cervical cancer from Madeleine et al’s 14 meta-analysis, and 7 HLAs were identified. This integrative approach resulted in identifying a subset of HLA alleles with both high worldwide prevalence and relevance to cervical cancer risk (Supplementary Table S2).
Subsequently, to evaluate the population-level applicability of the selected epitopes, population coverage analysis was performed. For MHC classes I and II, the selected epitopes showed a population coverage of 99.98%, indicating that nearly all individuals are predicted to present these epitopes. Moreover, the average hit value of 95.05 represented the mean number of epitope-HLA combinations per individual, and the PC90 value of 68.19 indicated the minimum number of combinations recognized by 90% of the population, supporting the broad genetic applicability of the proposed epitope set (Supplementary Figure S2).
Adjuvant identification and evaluation
Significant DEGs of the GSE63514 data set, which consists of 28 cervical squamous epithelial cancer samples and 24 normal cases, were retrieved using limma R-package calculations (based on P-value < 0.05 and |logFC| ⩾ 2). A total of 13154 DEGs encompassed 526 downregulated and 450 upregulated genes. The obtained gene set was subjected to enrichment analysis using integrated KEGG data. Genes were annotated with their functions, and highly significant enriched pathways were identified. The interleukin 17 (IL-17) signaling pathway showed one of the highest values (P-value = 2.35e-4) (Figure 2A).
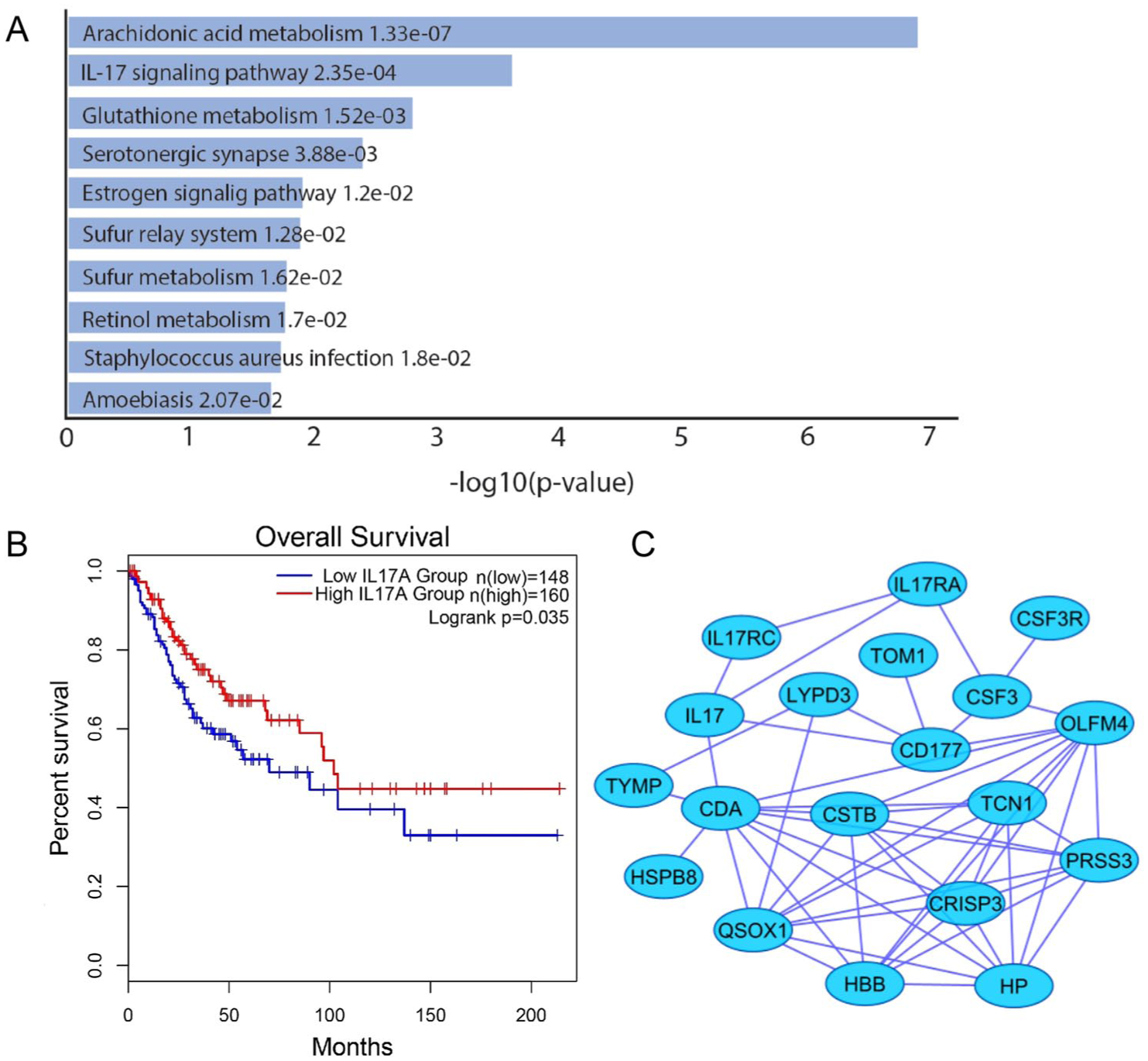
The outputs of the IL-17 pathway enrichment, survival analysis (based on the extent of IL-17 expression), and IL-17 clustered subnetwork. (A) Highly enriched pathways resulted from co-expressed DEG identification of the GSE63514 data set, demonstrating IL-17 signaling as second enriched axis; (B) The overall survival rate of patients with endocervical adenocarcinoma and squamous cell carcinoma of the cervix, where highly expressing group show an increased survival chance; (C) IL-17 hub genes network, which was extracted from GSE63514 data set using cytoHubba plugin and illustrated by STRING tool (DEG = differentially enriched genes).
Protein-protein interacting (PPI) network was modeled using the STRING database (with 408 nodes, 956 edges, and an average degree of 4.69). The significant enrichment of IL-17 signal transduction justified the rationale of its application as an appropriate adjuvant. To identify the receptors involved in signaling, the IL-17 network was extracted. The cytoHubba plugin of Cytoscape was employed to acquire all the associations of IL-17. The resulting subnetwork (with 20 nodes and 62 edges) exhibited an average degree of 6.2, which implies a higher extent of interconnection than the primary network (Figure 2C).
According to the Mantel-Cox test performed to assess the survival rates of patients with cervical squamous cell carcinoma, endocervical adenocarcinoma, and uterine carcinosarcoma over 200 months, higher levels of IL-17A (in 160 subjects) were significantly associated with higher proportions of survival (P-value = .035), compared with 148 subjects who expressed lower levels of IL-17A (Figure 2B).
Linear construction of vaccine candidates and evaluation of their physiochemical and biological attributes
Seven linear polypeptide constructs (A, B, C, D, E, F, and G) were designed based on different positions of the epitopes (Supplementary Table S3). To discover the most appropriate candidate, corresponding physiochemical properties, water solubility, allergenicity, and antigenicity were predicted (Supplementary Table S4). Among the candidates, structure D, which possessed an aliphatic index of 83.04 (a positive indicator of protein thermostability), was reported as a nonallergen compound, exhibited the highest solubility value (73%), and recorded the highest antigenicity (61%; by ANTIGENPro tool).
Homology 3D modeling of vaccine constructs, structure refinement, and B-cell conformational epitope prediction
The 3D models of linear vaccine sequences were predicted using the GalaxyTBM tool, and the GalaxyrRefine tool was employed to refine the structures. GalaxyRefine calculates the RMSD (root mean square deviation) and GDT (global distance test) indexes to score the modeled structures (lower values of RMSD imply less extent of alpha carbon (Cα) spatial movement; and resultantly, a more stable 3D design). However, higher GDT values indicate a higher possibility of appropriate Cα positioning (within a specific favorable spatial placement, thereby more construct stability). Construct D showed the lowest RMSD value (0.286) (Table 2).
Structural refinement and accuracy assessment of the modeled vaccine constructs; GalaxyRefine—predicted factors are reported for vaccine models to evaluate stereochemical reliability. Accordingly, lower RMSD values together with higher global distance test (GDT) values indicate structural quality.

The quality of the refined models was further validated using the ERRAT server (Supplementary Figure S1 and Table 2); accordingly, structure D had the highest overall quality factor (90.992%).
ProSA-Web scoring and Ramachandran plot-based analysis were further executed to analyze the refined models. The Z-score of -4.33, obtained from the ProSA-Web server, verified structure D based on all available NMR data and X-ray protein crystallography. The Ramachandran plot (of the Procheck server) also validated structure D based on the distribution of spatial residue positioning so that 94.8% of residues were placed within the most favored regions (Supplementary Figure S1).
ElliPro predicted potential conformational epitopes with affinity toward B-cells on the 3D conformations of modeled vaccines. Accordingly, 19 residues were identified as potent epitopes on model D (the 3D antigen constituted A204, A205, A206, K207, E208, A209, A210, A211, K212, P213, T214, L215, H216, E217, Y218, M219, L220, D221, and L222).
Immune response simulation analysis
Immune response simulations using the C-ImmSim server demonstrated biologically consistent activation of both innate and adaptive immune components following vaccination. The B-cell population remained elevated throughout the simulation period, with the emergence of memory B cells indicating the initiation of humoral immune memory (Figure 3A). A rapid increase in helper T (TH) cells was observed during the early phase of the simulation (days 0-12 approximately), alongside the development of TH memory cells (Figure 3B). Similarly, cytotoxic T (TC) cells exhibited early expansion followed by contraction (after day 10), reflecting antigen-driven cellular immune activation (Figure 3C). Analysis of macrophage (MA) and dendritic cell (DC) populations revealed active transitions among functional states, supporting effective antigen uptake and presentation (Figure 3D and E). Antigen kinetics showed a rapid clearance shortly after administration (about day 2 of simulation), consistent with immune recognition and processing (Figure 3F). The cytokine profile revealed a pronounced IFN-γ response with a clear early peak (around day 2-10) followed by gradual decline, while IL-2 and IL-4 exhibited only transient low-level increases and other cytokines remained near baseline (Figure 3G), indicating a predominantly IFN-γ-driven immune activation.

In silico immune response simulation of the designed multi-epitope vaccine; (A) B-cell populations show sustained activation with evidence of isotype involvement and memory formation following immunization; (B) Helper T (TH) cells exhibit early expansion and subsequent stabilization, accompanied by the emergence of memory cells; (C) Cytotoxic T (TC) cells display antigen-driven expansion followed by contraction and stabilization over time; (D) Macrophage populations transition from resting to active states, indicating efficient antigen uptake and processing; (E) Dendritic cell populations remain stable with active and presenting states supporting antigen presentation; (F) Antigen levels rapidly decline after administration, reflecting efficient immune clearance; (G) Cytokine responses are dominated by IFN-γ with only transient, low-level IL-2 and IL-4 responses.
Cross-docking analysis between adjuvant subunits of the vaccine and correspondent receptors
Using a flexible cross-docking analysis, protein-ligand interactions between the modeled vaccines and corresponding receptors were explored by the HADDOCK v2.4 software. HADDOCK v2.4 determines the optimal outcomes cluster by calculating RMSD and Z-value, then by predicting energy ranges for desolvation, Wander Valls, and electrostatic interactions (Table 3). In this study, a reference docking analysis was also carried out between bare IL-17A and IL-17 receptors (RA and RC); this was undertaken to assess the most correlated docking energies with the reference outputs by applying Pearson correlation (Table 3).
Cross-docking performance of IL-17A and modeled vaccine candidates (A-G) with IL-17 receptors; HADDOCK v2.4 output docking scores (mean ± SD or standard deviation), summarizes the relative binding quality and structural consistency of each vaccine-receptor complex compared with the IL-17A reference.

Consequently, compared with all harvested energies of modeled structures, docking outputs of the model D-IL-17RC construct displayed the highest correlation with the reference (IL-17A-IL-17RC), with a correlation value of .88. In addition, the correlation extent of the model D-IL-17RA complex (0.98) was ranked as the third highest amount (Figure 4A and B). The ligand-receptor interactions established in the binding pocket of model D-IL17RA/RC complexes are shown in Figure 4C and D. Binding site interaction analysis revealed stable hydrogen-bond networks at the vaccine—IL-17RA and vaccine—IL-17RC interfaces. The involvement of multiple polar and charged residues from both the vaccine construct and the receptors supports favorable docking correlations and structural stability observed for model D. The correspondent data are presented in Supplementary Table S5.

The results of cross-docking analysis of the vaccine constructs. (A) Correlations of the obtained IL-17RA docking analysis between the reference complex and other vaccine candidates (favorable, adequate, and nonfavorable correlation values are displayed with dark green, light green/red, and dark red, respectively), the correlation extent of the model D-IL-17RA complex (.98) was ranked as the third highest amount; (B) Correlations of the resulted IL-17RC docking analysis between the reference complex and other vaccine candidates, the correlation extent of the model D-IL-17RC complex (.88) was ranked as the highest amount; (C) model D (gray segment)-IL17RA (red segment) complex (whose active site has been magnified); (D) model D (gray segment)-IL17RC (red segment) complex.
Molecular Dynamics (MD) simulations of protein-protein interactions and subsequent calculations of MM/PBSA binding free energy
To better understand how modeled vaccines and the mentioned receptors interact, 2 sets of 500 ns-MD simulation runs were directed utilizing the favorable complexes (model D-IL-17RA and model D-IL-17RC), following the cross-docking step. After the EM step, which exerts a thermodynamical balance upon the system of each simulation, NPT (1 ns) and NVT (100 ps) ensembles were administered, which were then pursued by the main MD runs.
Both complexes’ RMSD values of Cα atoms were measured to obtain correspondent collective coordinates (Figure 5A and B). During the simulation time, both complexes underwent negligible fluctuations before reaching a stable status (which was recorded to be roughly 7 nm during both simulation runs) due to their efforts to obtain an optimum position. The radius of gyration (Rg), another indicator of conformational changes, was measured to assess the fluctuation in the overall shape and compactness of the complexes. After about 80 ns of the simulation time, and due to the proper establishment of inter-molecular and intra-molecular interactions, no considerable variation in Rg was reported, and the value was maintained at about 4 nm for both runs (Supplementary Figure S2). Analysis of the extent of fluctuation in the number of ligand-receptor hydrogen bonds showed a nearly constant number of hydrogen interactions in both complexes (below 500 and 600 interactions for D-IL-17RA and D-IL-17RC complexes, respectively). Data has been provided in supplementary Figure S2.
MM/PBSA analysis was employed to quantify and compare the binding affinities of the vaccine construct toward IL-17RA and IL-17RC receptors over the equilibrated phase of the simulations. The calculated energy components and total binding free energies for both complexes are summarized in Table 4. For both the vaccine-IL-17RA and vaccine-IL-17RC complexes, MM/PBSA analysis revealed consistently favorable binding free energies over the final 400 ns of simulation, indicating stable receptor engagement. Time-dependent energy profiles exhibited no significant deviation, indicating adequate convergence of the MM/PBSA estimates (Figure 5C and D).
MM/PBSA binding free-energy components for the vaccine-IL-17 receptor complexes; Mean ± standard deviation (kJ/mol) of van der Waals, electrostatic, polar solvation, nonpolar solvation, and total binding free energies calculated over the final 400 ns of molecular dynamics simulations (corresponding to the equilibrated and stable phase of the trajectories).


Structural stability of the vaccine-IL-17RA and RC complexes over a 500-ns molecular dynamics simulation(Figure 5A and B, respectively), RMSD profiles of the complexes show initial equilibration followed by convergence to a plateau after approximately 80 ns, indicating sustained structural stability for the remainder of the simulation; MM/PBSA total binding free energy profiles (kJ/mol) of the vaccine-IL-17RA and RC complexes (Figure 5C and D, respectively), calculated over the final 400 ns of the 500-ns simulations and corresponding to the period that the complexes reached structural stability (using 800 snapshots or one frame every 500 ps), indicating stable binding energetics over time.
In silico cloning of the vaccine
The vaccine construct (model D) was back-translated by EMBOSS backtrace to obtain the correlated DNA sequence, which was then inserted into the pQE30 plasmid and subjected to in silico cloning in E. coli K12. The JCat server optimized codon usage and the compatibility of the vaccine vector with the host bacterium. This implies an improvement in cytosine-guanine content and an increase in the adaptation index (CAI) to 0.96 (Table 5).
Codon optimization results to improve the compatibility of the designed vaccine construct to E. coli K12, demonstrating improved translational efficiency.

Using SnapGene software, the restriction sites of BamHI and HindIII enzymes were considered for the cloning sites’ 5’ and 3’ terminals, respectively (Figure 6). Finally, the NEBCutter online tool confirmed that the selected restriction enzymes are zero-cutters and do not interfere with the cloning process.
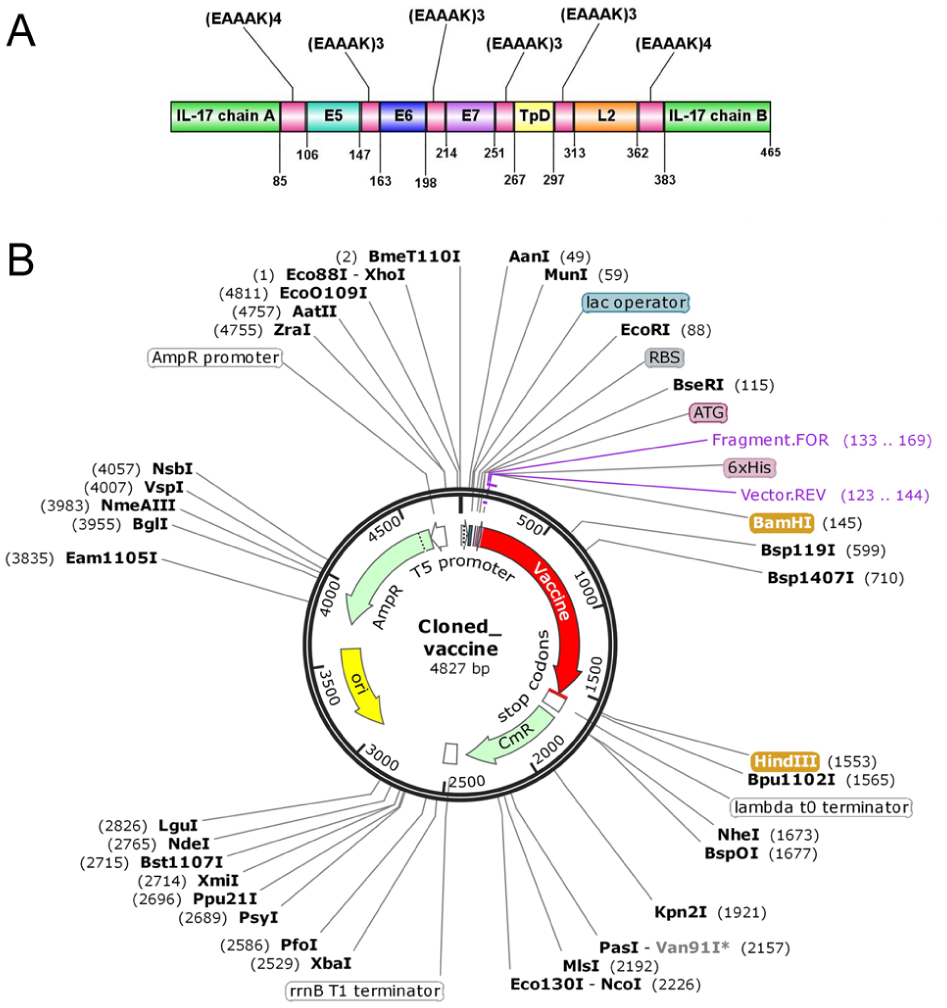
Schematics illustrations of the used sequences for in silico cloning. (A) A representation of the 465-residue-long vaccine (EAAAK oligomers are used as linkers, binding different selected epitopes and adjuvants together; epitopes are named according to their source protein and adjuvants employed, including IL-17 A and TpD sequence). (B) The inserted vaccine sequence (red) into the pQE30 plasmid with the restriction sites for BamHI and HindIII enzymes (highlighted in orange).
Discussion
Immunization by vaccination is a favorable prophylactic/therapeutic approach that assists the immune system in identifying and targeting pathogens and eliciting prolonged immunity. 4 Accordingly, applying peptides and epitopes is a rational method for vaccine development due to the simplified production procedure, chemical stability, lack of transmittable infections, and high safety.4,5 Multi-epitope vaccines could contain different protein segments from the pathogen (sometimes as overlapped epitopes) to induce humoral and/or cellular immunity. 52 This study implemented an immunoinformatics-based pipeline using systems vaccinology and proteomics to construct and design a multi-epitope vaccine against HPV-16 with both preventive and therapeutic potentials. HPV-16 was chosen as the most common HR-HPV type that causes cervical cancer, responsible alone for half of the cases. 2 Using in silico methods, CTLs, HTLs, and B-cell epitopes on HPV-16-E5, E6, E7, and L2 proteins were explored, and the best 19 epitopes, with favorable safety and immunogenicity profiles, were identified to construct the vaccine. Following vaccination, helper T lymphocytes (CD4+ cells or HTLs) and cytotoxic T lymphocytes (CD8+ cells or CTLs) contribute to the viral immunization process. Hence, the constructed vaccine should be composed of immunogenic sequences that stimulate both major histocompatibility complexes (MHCs).53,54 In epitope selection, the coverage of HLA alleles in various ethnic groups was also considered, as HLA diversity directly affects antigen processing and further presentation to B- and T-cells. 55 Accordingly, relevant HPV-16 E5, E6, and E7 epitopes were selected, achieving near-complete population coverage and broad epitope—HLA compatibility. 4
The phylogenic similarity between the selected epitopes of HPV-16 and the other 14 HR-HPVs revealed that HPV-16-E6 was similar to the E6 oncoprotein of HPV-35, -33, -58, -31, and -73, respectively; as for HPV-16-E7, the similarity was reported for E7 of HPV-31 and -35. Given that HPV-31, -33, -35, and -58 are all oncogenic HR-HPV types, and HPV-73 is a probably-oncogenic HPV type, 4 the therapeutic epitopes of HPV-16 may cover 6 HR-HPV types, which could be regarded as a great approach. Subsequently, HPV-16-L2 was shown to be phylogenetically similar to the L2-capsid protein of HPV-35, -31, -33, and -58. Thus, the prophylactic effect of the vaccine could potentially also work against 4 HR-HPV types other than HPV-16.
Notably, compared with currently available vaccines such as Cervarix (targeting HPV-16 and -18), Gardasil (targeting HPV-6, -11, -16, and -18), and Gardasil 9 (extending the coverage spectrum of the previous product to HPV-31, -33, -45, -52, and -58), the proposed vaccine construct targets cross-conserved epitopes, and thus targets 6-subtypes (including HPV-16, -31, -33, -35, -58, and -73). This demonstrates promising potential to offer broader protection against additional HR-HPV types, which are not covered by some of the existing commercial vaccines. 7
Compared with previous studies, we attempted to implement HPV-16 E5 subunit in the designation of our construct . This subunit could play a significant role in conferring preventive and therapeutic properties since it has been shown that E5 is expressed during the early stages of HPV infections and plays a vital role in altering cellular protein pathways and activity, mainly in growth factor pathways. 4 Ultimately, the use of E5 protein possibly provides a massive opportunity to help prevent and suppress precancerous legions and more progressive carcinomas. 56
After infection with HR-HPVs (mostly HPV-16 and -18), stimulation of cytokine production in the malignant microenvironments is compromised. For instance, immune evasion behaviors of HR-HPVs impede IL-17 induction. This implies HPV-E6 and -E7-exerted negative regulation of TLR9, or Toll-like receptor 9 (leading to compromised innate immune response) and HPV-E5-attributed subregulation of MHC expression (which could affect CD4+ T cell activation by MHC-II-dysregulated antigen presentation). 57
Considering the low immunogenicity of peptide vaccines, which results mainly from their lack of complex conformational complexities, adjuvants are an essential part of polypeptide vaccines. 58 Adjuvants prolong, modulate, and enhance immune responses toward therapeutic/prophylactic vaccines. Furthermore, they reduce the demanded antigen concentration and number of booster doses, which renders the vaccine cost-effective. 8 These molecules are generally considered in vaccine design, albeit some studies have not included them within their sequences, as synthetic long peptides themselves possess intrinsic agonistic attributes.59,60 However, there are multiple collective findings to advance indications for implementing adjuvants; as they facilitate peptide presentation to MHC molecules via transducing key signals to activate adaptive immune system (eg, CTLs, HTLs, gdT-cells, and NK T-cells). 8 In our study, 2 adjuvants were employed, including TpD (a chimeric MHC-II peptide comprised of epitopes from diphtheria and tetanus toxoid) 61 and IL-17. The selection of IL-17A as an adjuvant in this study was guided by a systems vaccinology rationale, rather than empirical or conventional adjuvant choice. Similar to recent immunoinformatics-based cancer vaccine studies that use transcriptomic profiling and pathway enrichment to justify immune targets.62-64 Moreover, and as demonstrated in other multi-epitope vaccine frameworks, rationally selected adjuvants are incorporated to enhance antigen presentation, T-cell priming, and antitumor immunity in contexts of immune evasion.62-64 In other words, this study introduces an new insight into IL-17A application as a vaccine adjuvant through systems vaccinology and based on the dynamic of gene interactions and their expression response to perturbations (in malignancies).55,65
The DEG coexpression analysis of the GSE63514 microarray data set identified IL-17A transductions as the second most functionally enriched signaling pathway, which rationalized the application of IL-17 as the adjuvant of the vaccine sequence. “Omics” methods and computational approaches (such as systems vaccinology) are used extensively to predict immunogenicity and detect molecular signatures and their corresponding adaptive immune responses.66-68 Therefore, microarray data from Boon and colleagues’ study 31 was employed to identify critical genes (whose expression profile changed the most under malignant circumstances) and conduct molecular network modeling.65,69
According to the coexpression screening of DEGs, IL-17 transcriptional alteration significantly correlates with cervical tissue remodeling.70,71 By activating/recruiting HTLs, CTLs, neutrophils, and natural killer (NK) cells to the malignant tissues, IL-17 exerts anti-tumor properties, which justifies the use of this cytokine as a potential adjuvant. For instance, NK cells are induced to synthesize cytotoxic molecules such as interferon-gamma (IFN-γ), myeloperoxidase (MPO), and reactive oxygen species (ROS) when treated by IL-17.72,73
However, IL-17 is reported to activate NF-κB, JAK2/STAT3, and PI3 K/Akt signaling pathways. Therefore, cell proliferation is promoted, and apoptosis is inhibited when STAT3 is translocated and Akt is activated. 74 In addition, when fibroblasts or stromal cells are induced by IL-17, the expression of growth factors (eg, granulocyte colony-stimulating factor [G-CSF] and vascular endothelial growth factor or [VEGF]) is upregulated, resulting in angiogenesis.57,70
Despite the tumorigenic features, the association between tumor immunopathology and IL-17 functionality is highly correlated to the context of the tissue microenvironment. In esophageal squamous cell carcinoma, infiltration of IL-17-producing cells recruited CD8+ T cells, B cells, and NK cells, and thus, induced protective immunity. 73 In colorectal cancer, IL-17-secreting cells promoted antitumor immunity by the recruitment of MPO-producing neutrophils. 75 In ovarian cancer, increased IL-17 production improved the survival rate. Moreover, the extent of IL-17 was shown to be positively correlated with the presence of IFN-γ+ CD8+ and IFN-γ+ CD+ T-cells within the tumor microenvironment. 76 In tumor tissues, infiltration of antitumor neutrophils (known as N1-phenotype) is recruited by IL-17. N1-neutrophils are generated when TGF-β is depleted and induce multiple immunostimulatory cytokines (eg, TNF-α, macrophage inflammatory protein-1α [or MIP-1α], hydrogen peroxide, and nitric oxide) with increased potential to eliminate tumor cells in vivo and in vitro. 77
Various in vitro studies indicated that exogenous IL-17 had no direct impact on tumor cell growth.78-80 Regarding in vivo findings, IL-17 administration in mice has demonstrated a favorable safety profile, with stable hematological parameters and no significant adverse changes in cell populations observed. 81
Reportedly, exogenous IL-17 transduces fibrosarcoma cells to elicit antitumor immunity by inducing the expression of MHC class I and class II tumor-specific antigens. 79 Furthermore, increased expression of allostimulatory capacity and MHC-II antigens have been reported to indicate exogenous IL-17-promoted maturation of dendritic cell progenitors, which results in the improvement of T cell priming. 82 Notably, the time scale of IL-17 induction could be a determining factor, as Takeuchi et al’s 83 findings on the immune response toward BCG vaccine administration for bladder cancer patients revealed that a short duration of IL-17 production might not promote tumor progression. Thus, human IL-17A (chains A and B) were used in our construct as adjuvants besides TpD. The homo/hetero dimers of IL-17 consist of 2 monomers connected through a cysteine-derived disulfide bond. IL-17A is the fundamental member of the IL-17 family, which is comprised of IL-17A, B, C, D, E, and F. IL-17 signals through a complex of IL-17 receptor (IL-17R) subunits.70,71
IL-17R is a family of single-pass transmembrane proteins with 5 members (ie, IL-17RA, IL-17RB, IL-17RC, IL-17RD, and IL-17RE). IL-17 interacts with IL-17R complex with at least 1 IL-17RC and 2 IL-17RA subunits (rending IL-17RA and RC is highly critical for the interaction). 72 Furthermore, based on the transcriptomic profiling of patients with cervical squamous cell carcinoma (Figure 2), the IL-17 signaling pathway was found to involve 2 key receptors (ie, IL-17RA and IL-17RC). This further provided a rationale for conducting cross-docking simulation studies focused on the aforementioned receptors.
Thus, cross-docking simulation analysis was conducted between the IL-17 subunit of the vaccine and IL-17RA and RC since IL-17 binds to a receptor comprised of at least IL-17RA or RC. 72
EAAAK oligomers were used as linkers to bind the epitopes and adjuvants. According to Shahab et al, 84 who conjugated fragments of HPV-16-L2 with 50-s ribosomal protein from Mycobacterium tuberculosis (as an adjuvant), appropriate selection of linkers can also enhance immunogenicity. In addition, EAAAK is a rigid α-helical sequence that spatially separates functional domains, preserving the structural and immunological independence of epitopes and adjuvant components. Its effectiveness has been demonstrated in multi-epitope vaccine studies through improved structural stability and validation in cancer models, 85 enhanced construct stability in viral vaccines, 86 and its function as a stable domain spacer via intramolecular hydrogen bonding, 87 supporting its use in our construct. Different studies have aimed to combat HPV by designing various vaccines using reverse vaccinology approaches. For instance, one of our attempts was focused on developing a prophylactic vaccine, 2 which consisted of HPV-16 -L2 and a complex adjuvant, including flagellin (a TLR5 agonist), a short synthetic TLR4 agonist, and 2 universal HTL agonists (TpD and PADRE). The in vivo investigations on this vaccine showed its efficiency in the induction of immune responses. 58 Therapeutic vaccines have also been investigated in different studies. 58 In this study, we aim to induce prophylaxis and elicit therapeutic attributes by administrating fragments of HPV-16-L2 VLPs. Furthermore, compared with our previous prophylactic/therapeutic vaccine that exploited a TLR4 agonist and HPV-16-L2, E6, and E7 epitopes. 5 Our current design also includes the E5 sequence to increase therapeutic efficacy.
In this study, the analysis of the physiochemical properties of all candidate constructs and docking results identified the most favorable modeled vaccine (model D). The immune response simulations further indicated robust activation of innate and adaptive immune responses, including memory cell formation and a CD4+ biased cytokine profile. The selected vaccine construct was then subjected to 2 sets of MD simulations, and the stabilized establishment of intermolecular and intramolecular interactions was demonstrated (evaluated by measurement of RMSD, H-bond fluctuations, and Rg values). The reported low values of RMSD and a steady number of H-bonds indicated no significant conformational folding/unfoldings during a simulation of 160 ns. The constant Rg measure specified a maintained mass-weighted RMSD of the collection of atoms from the center of complexes (D-IL-17RA and D-IL-17RC). MM/PBSA analysis indicated stable and favorable binding of the vaccine construct to both IL-17RA and IL-17RC, with intermolecular interactions effectively overcoming unfavorable solvation effects. Energy profiles resulted from 800 snapshots over the final 400 ns showed no significant systematic change, supporting the robustness and reliability of the binding free energy estimates.
Conclusion
The prophylactic and therapeutic vaccine candidate designed in this study, which contains epitopes from L2 and E5, E6, and E7 proteins of HPV-16, was found to be a promising construct through the performed in silico analyses. The designed vaccine could potentially work against HPV-16 and multiple other HR-HPV types. Adding epitopes of HPV-18 in the future could further expand the coverage of the vaccine against important oncogenic HPV types. Based on the systems biology approaches carried out in this study, it was shown that IL-17 could serve as a proper vaccine adjuvant with desired immunological properties. The appropriate interaction of IL-17 with its receptor was verified by docking and MD studies.
Of note, while computational strategies enable rapid, cost-effective vaccine design, they are limited by the outputs built upon predictive algorithms. These include potential inaccuracies in B- and T-cell epitope prediction, conformational modeling limitations (eg, discontinuous epitopes), and the inability to account for complex in vivo immunodynamics such as cytokine storms or immune tolerance mechanisms.
Resultantly, future extensive experiments (eg, in vitro and in vivo studies) are needed to validate the efficacy and safety of this vaccine. Particular attention must be given to ensuring peptide stability -while minimizing adverse effects- and the development of efficient delivery systems. In addition, the variability in host MHC profiles, the emergence of viral variants that may influence immune responses, and regulatory and ethical considerations related to preclinical studies are of high importance regarding the challenges in transitioning from in silico settings toward translational application. Notably, compared with existing HPV vaccines and prior immunoinformatics-based designs, the present construct integrates both early and late viral antigens together with a systems vaccinology guided cytokine adjuvant, enabling broader subtype coverage and combined prophylactic and therapeutic potential. Future efforts will therefore focus on experimental validation of immune activation, controlled assessment of IL-17A-mediated responses, and preclinical evaluation of immunogenicity and safety to support further translational development.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322261437247 – Supplemental material for Systems Vaccinology-Integrated Proteomics to Develop a Novel Prophylactic and Therapeutic Vaccine Against Human Papillomavirus 16
Supplemental material, sj-docx-1-bbi-10.1177_11779322261437247 for Systems Vaccinology-Integrated Proteomics to Develop a Novel Prophylactic and Therapeutic Vaccine Against Human Papillomavirus 16 by Shirin Fathi, Aida Solhjoo, Ashkan Bagheri, Amirhossein Sakhteman, Younes Ghasemi and Manica Negahdaripour in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
This study was a part of the Pharm.D. thesis of Shirin Fathi.
Ethical considerations and consent for participation/publication
Not applicable. This study did not include any human participants or animals.
Author contributions
Shirin Fathi: Writing—original draft; Data curation; Visualization; Investigation; Validation; Writing—review & editing.
Aida Solhjoo: Data curation; Validation.
Ashkan Bagheri: Writing—original draft.
Amirhossein Sakhteman: Methodology.
Younes Ghasemi: Conceptualization.
Manica Negahdaripour: Conceptualization; Writing—review & editing.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
All data supporting the findings of this study are openly shared and also available from the corresponding author upon request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.