
Abstract
Petroleum hydrocarbon pollution is an escalating global issue, particularly in developing countries, where it has attracted significant attention from researchers focusing on bioremediation, monitoring and sustainability. This study utilised metagenomics to investigate the bacterial community’s response in polluted soil undergoing field-scale biopile treatment, with chicken droppings as a nutrient source. Hydrocarbon concentrations were monitored over a 90-day remediation period using the Fourier transform infrared (FTIR) spectrometry technique. Molecular and bioinformatic analyses were conducted to track the dynamics of bacterial species, their abundance and functional roles during the bioremediation process. The initial total petroleum hydrocarbon (TPH) concentration of 446 945 ppm was first reduced to 80 332 ppm through dilution. Following a 90-day bioremediation process using poultry waste, the level further decreased to 5326 ppm, representing a 93.37% reduction. In the metagenomic analysis, a total of 26 736 reads were obtained, averaging 6684 counts per sample. In addition, the study identified diverse bacterial metagenomes, including well-established hydrocarbon-degrading bacteria from Proteobacteria, Firmicutes, Acidobacteria and Actinobacteria phyla, and species previously not reported as hydrocarbon-degrading. Biomarkers associated with hydrocarbon metabolisms, such as aromatic dioxygenases, alkane-1-monooxygenase and methanol oxidation pathways, were identified. A significant decrease in the relative abundance of bacterial genera in heavily polluted soil was observed, alongside an increased presence of Caballeronia, Paraburkholderia and Fontibacillus genera. These findings indicate that chicken droppings contribute 0.30% to the reduction of TPH in the biopiling remediation technique used for treating heavily contaminated soil. A comparative assessment of hydrocarbon attenuation in nutrient-amended vs unamended soils indicates that a 3-month remediation timeframe is insufficient to achieve optimal bioremediation outcomes. However, the TPH reduction in unamended treatment highlights the intrinsic natural attenuation capacity of the impacted soil matrix, attributable to indigenous microbial consortia and prevailing environmental conditions.
Keywords
Introduction
Hydrocarbon pollution is a global issue, prompting international, regional and national bodies to establish benchmarks using total petroleum hydrocarbons (TPHs) as a key indicator. Organisations such as the United Nations Environment Programme (UNEP) and the United States Environmental Protection Agency (US EPA) provide general guidance for assessing and remediating petroleum-contaminated sites, recommending a technical benchmark of 1000 mg/kg for TPH.1,2 In Nigeria, the Department of Petroleum Resources (DPR), through its Environmental Guidelines and Standards for the Petroleum Industry in Nigeria (EGASPIN), sets a safe limit of 50 mg/kg and an intervention threshold of 5000 mg/kg. 3 However, in developing countries such as Nigeria, regulatory standards are often overshadowed by the economic priorities of oil exploration. This has led to various forms of hydrocarbon pollution, including accidental discharges, pipeline ruptures, leaks, deliberate discharges, vandalism and artisanal refining activities. 4 These activities significantly impact air, water and soil ecosystems. The resulting polluted conditions reduce biodiversity and biomass, compromise ecosystem services and lead to the bioaccumulation of toxic substances in animals and humans. These effects weaken the immune system, cause genetic diseases and increase cancer risks. 5 Addressing these negative impacts requires remediation schemes such as physical, chemical and biological processes. Although it takes longer, the biological process (bioremediation) has a comparative advantage due to its many benefits, such as sustainability, low technological requirements, cost-effectiveness, minimal secondary pollutants, ease of knowledge transfer, eco-friendliness and high compatibility index. 6 Bioremediation is often used alongside physical and chemical methods. This includes techniques such as biostimulation, bioaugmentation and bioattenuation. A unique feature of soil bioremediation is its ability to leverage organic waste, which provides essential nutrients, supports a consortium of hydrocarbon-degrading microorganisms and improves soil handling properties. This valorisation of organic waste for hydrocarbon remediation promotes the concept of ‘waste-for-remediation’. Such remediated soil can also become activated soil, which may be used to remediate other contaminated areas. 7 The success of hydrocarbon remediation is typically measured by the reduction in TPH, assessed through chromatography, spectrometry and metagenomic analyses. Metagenomics monitors the composition, structure and diversity of bacteria, fungi and algae, providing valuable insights into the microbial remediation process.
Metagenomics is a rapidly expanding field dedicated to uncovering the true diversity of microbes, their interactions, functions and evolution across diverse ecosystems. Powered by next-generation sequencing (NGS), this field has moved beyond traditional cloning methods, enabling advances in comparative metagenomics. 8 High-throughput technological advancements have revolutionised metagenomics, allowing the rapid generation of vast datasets. As a culture-independent, sequencing-based omics tool, metagenomics examines community DNA, offering reliable insights into the presence and activities of microorganisms. Metagenomic analysis can be broadly categorised into sequence-based and function-based approaches. This method is critical for genome assembly, comparing microbial communities and predicting metabolic pathways and gene functions. 9 Functional metagenomics investigates gene functions by isolating DNA from environmental samples to study the roles of encoded proteins. 10 This approach involves cloning DNA fragments, expressing them in a suitable host and assessing enzymatic activities. 11 It is instrumental in discovering novel genes and understanding metabolic and functional diversity. Therefore, metagenomics is a dynamic and powerful tool that provides comprehensive insights into the unseen world of microorganisms, their genetic makeup and functional capabilities. With continuous advancements in NGS technologies, metagenomics is poised to revolutionise our understanding of microbial communities and their roles in various ecosystems.
Malla et al 12 highlighted the unparalleled value of metagenomics in bioremediation studies, emphasising that it surpasses other omics approaches in providing detailed insights into degradation processes. They argued that metagenomics significantly advances our understanding of how microorganisms acquire the ability to detoxify xenobiotic substances through in situ or ex situ treatments. Furthermore, it facilitates the discovery of key microbial activities, elucidates the role of community structures in pollutant mineralisation and offers extensive gene databases for developing microbial strains tailored to specific bioremediation efforts. Microbial ecologists regard metagenomics-driven monitoring of bioremediation as indispensable. Ejaz et al 13 supported this perspective, highlighting that metagenomics uncovers numerous novel molecules with significant functional applications, particularly enzymes with enhanced properties compared to those identified from cultured microorganisms. Similarly, Abdoli et al 14 demonstrated the utility of metagenomics in identifying genes involved in hydrocarbon degradation pathways and the type VI secretory system (T6SS), providing critical insights into petroleum hydrocarbon-contaminated soils. In addition, Anju et al 15 demonstrated that metagenomics offers a comprehensive understanding of complex microbiomes and serves as a holistic indicator of ecosystem responses to pollutants. By leveraging bioinformatics, metagenomics extracts valuable information on DNA, RNA, metabolites and proteins, enabling a deep understanding of microbiome composition, functional activities and community responses to pollutants at the molecular level. Despite its vast potential, much of the progress in metagenomics research is driven by studies conducted in advanced countries and top universities, highlighting a need for broader global participation.
In developing countries like Nigeria, there is an insufficient information on the molecular aspects of pollution and bioremediation. To address this gap, this research aimed to enhance the limited knowledge of hydrocarbon bioremediation metagenomics in the Niger Delta. The primary objective of this study is to investigate the changes in bacterial communities alongside hydrocarbon attenuation during the bioremediation process, using chicken droppings as a nutrient source to stimulate hydrocarbon-degrading bacteria. Our findings showed a 93.37% reduction in hydrocarbon concentration, with chicken droppings accounting for a 0.30% reduction. The Amplicon Sequence Variants (ASVs) of Proteobacteria, Actinobacteria, Firmicutes and Acidobacteria dominated the analysed samples. The observed ASV count represents the relative abundance of taxa, as is expected in metagenomic studies. The application of chicken droppings increased the abundance of Proteobacteria and Firmicutes, enhanced microbial diversity and amplified the relative abundance of hydrocarbon-degrading genera such as Caballeronia and Methylocystis, particularly in the heavily polluted samples. In addition, there was a significant increase in the expression of enzymes associated with aromatic compound degradation pathways, particularly catechol and protocatechuate. This study underscores the potential of using residual organic materials, such as chicken droppings, to remediate hydrocarbon-contaminated soil effectively, simultaneously promoting a ‘waste-for-solution’ approach in environmental remediation and sustainability initiatives. Interestingly, the study found no significant difference in hydrocarbon attenuation between nutrient-amended and unamended soils, implying that the polluted soil may possess its own self-remediation capabilities.
Materials and Methods
Site description
The research was conducted on polluted agricultural land in Ngia Ama (latitude 4°47’42’’ N, longitude 6°51’45’’ E) in the Tombia Kingdom, Nigeria. This site is surrounded by mangroves and creeks with moderate lowlands with a temperature range of between 27 and 30°C and impacted by illegal refining activities over the past 7 years. Sections of the site containing oil reservoirs are noticeably contaminated with crude oil and hence categorised as heavily polluted soil (HPS). In contrast, areas further from these heavily polluted sections are classified as mildly polluted soil.
Experimental design
The HPS was blended with mildly polluted soil to dilute the high concentration of contaminants. The mixing procedure was carried out systematically. First, vegetation on the mildly polluted soil was removed. A 1.5 ft × 2 ft section of the HPS was excavated to a depth of 3 ft. A shovel-load of the recovered HPS was then spread over ca. 2 ft2 area of the mildly polluted soil (15 ft × 15 ft) and left to dry for 2 days and then crushed. Thereafter, both soils were thoroughly mixed using a shovel and pooled together. The field-scale bioremediation was conducted during the dry season to mitigate the potential leaching and dispersal of contaminants caused by precipitation-driven runoff. The resulting mixture was used to form 51 ridges measuring 3 ft ×1 ft × 0.3 ft. These ridges were divided into 2 halves: 26 were treated with chicken droppings (MAS-1) at a ratio of 4 parts mixed soil to 1 part chicken droppings (measured by wheelbarrow volume, with the chicken waste making up 25%), 16 while the other 25 ridges received no nutrient treatment. Figure 1 shows a visual representation. The poultry droppings were subjected to aerobic composting with wood chips in an exposed, sunlit environment for a duration of 30 days, facilitating their complete maturation into nutrient-rich manure.

Process flow of the experimental procedure during the field bioremediation process.
This procedure was conducted at an off-site location to ensure controlled decomposition. Over 90 days, the ridges were tilled and watered using agrotechnical methods at 7-day intervals. An unpolluted soil sample (UPS) was collected 2 m away from the affected site. Weekly composite samples were taken from both treated and untreated ridges, including the amended sample on day 90 (MAS-90) and the unamended sample on day 90 (MSS-90). Systematic random sampling was employed to collect composite samples. Samples were taken from the right end of all the ridges, combined, thoroughly mixed, and a representative portion was collected. Sampling depth is ⩽ 30 cm. The same procedure was repeated for the left end and the middle section of all the ridges. For each sampling event, 6 composite samples were collected: 3 from the amended soil and 3 from the unamended soil. Each representative sample (of HPS, MAS-1, MSS-90, MAS-90 and UPS) was divided into 2 parts, with 1 portion stored at 4°C for physicochemical and microbiological analyses, and the other at −20°C for molecular studies, following the methods of Hallmaier-Wacker et al 17 and Ahannach et al. 18
Physicochemical analysis of soil
Total nitrogen was conducted according to Zhang et al. 19 Total phosphorus was ascertained using the inductively coupled plasma optical emission spectroscopy (ICP-OES), PerkinElmer’s Avio Max Series, by Hamilton et al 20 protocol. Soil pH was carried out by adding 10 g of each sample to 100 mL of clean beakers. Deionised water (20 mL) was added, and the suspension was thoroughly stirred with a glass rod for 30 minutes to obtain a homogeneous mixture. After that, a calibrated pH metre (pH tester 20) was dipped into the beaker containing the suspension, and the pH value was recorded after a steady reading. The average pH value was taken from triplicate readings.
Determination of total petroleum hydrocarbons
First, 2 g of soil sample was heated to 50°C and thoroughly crushed. Then, 10 mL of dichloromethane (from Sigma Aldrich, USA) was added to the crushed soil, and the mixture was vigorously shaken. It was centrifuged at 300g for 10 minutes to separate the soil, following the method by Minai-Tehrani and Herfatmanesh. 21 The solvent phase was carefully removed through careful filtering. The extracted crude oil was stirred with a magnetic stirrer for 5 minutes and then filtered using Whatman No. 42 filter paper. 22 The mixture was then evaporated in a water bath until the volume was reduced to 1 mL, as described by Sari et al. 23 This concentrated extract was used for TPH analysis using a Fourier transform infrared (FTIR) spectrometer (Thermo Scientific Nicolet iS5) according to Hasan et al. 24 Absorbance was recorded at 2930 cm−1 wavenumber for the infrared spectra picking of TPH since the determination relied on a fixed-wavelength instrument responding to C-H bond stretching near 2850 to 2950. 25 In addition, a procedural blank was determined by performing the extraction and clean-up procedures with glass beads instead of a soil sample, following Sari et al. 23
Statistical significance analysis of total petroleum hydrocarbon
To assess the statistical significance of the hydrocarbon concentration data for 4 experimental groups (MSS-1, MSS-90, MAS-1 and MAS-90) and 2 controls (UPS-1 and HPS-1). The 6 data are first organised into columns within an Excel spreadsheet. The Data Analysis ToolPak is then utilised to perform a 1-way analysis of variance (ANOVA), selecting ‘ANOVA: Single Factor’, defining the appropriate data range and setting the significance level (typically α = 0.05). If the ANOVA result is significant (P < .05), indicating differences among groups, post hoc test is conducted using t-test (2-sample assuming equal variances) pairwise comparisons. Given the multiple comparisons involved, the Bonferroni correction is applied to adjust the significance threshold, ensuring control over the group-wise error rate. Finally, adjusted P-values are calculated and compared to the corrected α to determine statistically significant differences between groups, ensuring robust and reliable inferential outcomes.
Metagenomic analysis based on 16S
DNA extraction
DNA was extracted using the Zymo Research (ZR) Fungi/Bacteria DNA MiniPrep kit (California, USA), supplied by Inqaba Biotec (South Africa), following the manufacturer’s protocol with 2 modifications. Soil samples stored at −20°C were equilibrated to room temperature for 1 hour before extraction. In total, 1 g of soil was resuspended in 750 µL of BashingBead Buffer and transferred to a ZR BashingBead Lysis Tube. Samples were homogenised in a bead beater at maximum speed for 20 minutes (instead of 5 minutes). Following lysis, tubes were centrifuged at 10 000g for 2 minutes (instead of 1 minute). A 200-µL aliquot of the supernatant was transferred to a Zymo-Spin III-F Filter in a collection tube and centrifuged (10 000g, 1 minute). The filter was discarded, and 600 µL of Genomic Lysis Buffer was added to the flow-through, mixed thoroughly and loaded onto a Zymo-Spin IICR Column in a fresh collection tube. The column was centrifuged at 10 000g for 1 minute and transferred to a new collection tube. The column was washed sequentially with 200 µL DNA Pre-Wash Buffer and 500 µL g-DNA Wash Buffer, each followed by centrifugation at 10 000g for 1 minute. It was then placed into a clean 1.5-mL microcentrifuge tube (improvised). DNA was eluted with 35 to 100 µL DNA Elution Buffer and recovered by centrifugation (10 000g, 1 minute). Purified DNA was stored at −20°C for long-term use. The modifications were informed by Shen. 26 A single soil sample was collected per experimental group for metagenomic sequencing to ensure representative microbial profiling while maintaining analytical feasibility.
Polymerase chain reaction
The polymerase chain reaction (PCR) analysis using 341F (CCTACGGGNGGCWGCAG) and 785R (GACTACHVGGGTATCTAATCC) primers was designed to amplify the V3-V4 hypervariable region of bacterial 16S rRNA genes, facilitating microbial community profiling. The PCR reaction was prepared in a 50-µL total volume, incorporating 100 ng of template DNA, 0.2 µM of each primer, 200 µM dNTPs, 1× PCR buffer with MgCl2 (1.5-2.5 mM), 1 unit of Taq polymerase and PCR-grade water to adjust the volume. The thermal cycling conditions started with initial denaturation at 95°C for 5 minutes, followed by 35 cycles of denaturation at 95°C (30 seconds), annealing at 60°C (30 seconds) and extension at 72°C (45 seconds), concluding with final extension at 72°C for 5 minutes and a hold at 4°C. Post-PCR, agarose gel electrophoresis (1% gel) was conducted to confirm amplification, and purified amplicons were processed using bead-based or column purification methods to remove contaminants. The processed samples were subsequently prepared for sequencing using the Illumina MiSeq platforms, ensuring high-resolution taxonomic classification of bacterial communities. 27 All the molecular analysis including sequencing was done at the University of South Africa, Johannesburg, South Africa.
Bioinformatics analysis
Paired-end reads obtained from the sequencing facility were demultiplexed and quality-checked using FastQC software version 0.11.5. Subsequently, Trimmomatic software (version 0.38) developed by Bolger et al 28 was used to quality-trim the paired reads, including removing Illumina barcodes and eliminating reads with an average quality score (Phred Q score) lower than 20. The quality-filtered paired reads were further analysed using Qualitative Insights into Microbial Ecology (QIIME2) as described by Bolyen et al. 29 To merge the paired-end sequences into full-length sequences and eliminate chimaeras, we used the DADA2 denoiser version 1.14. 30 Amplicon Sequence Variants were clustered from similar sequences at a 97% similarity threshold using USEARCH version 7. 31 The taxonomic classification of these clustered ASVs was performed against the SILVA reference database version 138. 32 With a high number of unclassified reads, we classified the representative sequences in FASTA format using rrnDB Estimate (https://rrndb.umms.med.umich.edu/estimate/). The resulting ASV table, taxonomy table and sample metadata were imported into RStudio for visualisation following the tutorial for microbiome analysis in R. 33
For further analysis, we used the ASV table in BIOM format, and the representative sequences obtained from QIIME2 as input for visualisation, following the tutorial for the Nephele platform from the National Institute of Allergy and Infectious Diseases (NAID) Office of Cyber Infrastructure and Computational Biology (OCICB) in Bethesda, MD. The goal was to predict metabolic functions using PICRUSt2. In addition, we visualised the PICRUSt2 output using STAMP version 2.8 and MicrobiomeAnalyst, which provides comprehensive statistical, functional and integrative analysis of microbiome data. 34 The resulting ASV table was rarefied to an even depth sequence before computing alpha diversity (pairwise test with Wilcoxon rank-sum test, corrected by False Discovery Rate (FDR) method for the number of observed, Chao 1, Abundance-based Coverage Estimator (ACE), Shannon and Simpson) and beta diversity (using Bray-Curtis and binary Jaccard distance metrics). For differential abundance analysis (DAA), we used the ALDEx2 package (Swift et al, 35 Lin and Peddada, 36 ). The EC and pathway output from PICRUSt2 were visualised using STAMP. Specific enzymes and pathways were selected from the EC and pathway outputs, and the profile bar (P-value filter < .05) and G-test (W/Yates’) statistical test were employed for comparisons between different samples.
Results
Soil total petroleum hydrocarbon and physicochemical properties
Infrared spectrometry analysis revealed surface TPH concentrations of 446 945 ppm in the HPS, 92 693 ppm in mixed soil without nutrient (MSS-1), 80 332 ppm in the mixed soil with nutrients (MAS-1) and 25.33 ppm in the control soil (UPS), as shown in Table 1. The dilution percentage of MSS-1 in TPH concentration was 79.26%, while MAS-1 had a dilution percentage of 82.03%. After a 90-day remediation period, the TPH concentration decreased to 5326 ppm (93.37%) in the nutrient-treated sample (MAS-90) and 5862 ppm (93.67%) in the unamended soil sample (MSS-90). The percentage difference in residual TPH concentrations between MAS-90 and MSS-90 was 0.30%, attributed to the influence of organic fertiliser on TPH attenuation. This result reflects the cumulative effect of dilution, vaporisation and biodegradation, with dilution having the greatest impact due to the adopted remediation method. The statistical significance analysis generated an output of 15 Bonferroni corrected P-values at .0083. The TPH concentration of the heavily polluted sample showed significance against all the other samples. The unpolluted control sample (UPS-1) showed significance to all the other samples except in MSS-1, which is not expected. MSS-1 Vs MSS-90 showed a P-value of .1414, while MAS-1 vs MAS-90 pair showed a P-value of 1.2 × 10−8. The 2 terminal residual hydrocarbon, as expected, showed no statistical significance. Overall, residual organic matter (ie, chicken droppings) showed no statistically significant difference compared to the unamended soil. Total nitrogen ranged from 22.7 mg/kg in the HPS to 583 mg/kg in the nutrient-amended soil, while total phosphorus ranged from 16.3 mg/kg in the HPS to 188 mg/kg in the amended soil.
Physicochemical properties of the sampled soil.

HPS: heavily polluted soil sample; MAS: manure (chicken droppings) amended mixed soil sample; MAS-90: amended soil sample after treatment; MSS: unamended mixed soil sample; MSS-90: unamended soil sample after treatment; UPS: unpolluted soil sample. The accompanying hyphens 1 and 90 indicate the sampling day. For example, MSS-1 means unamended mixed soil sampled on day 1, while MSS-90 means unamended mixed soil sampled on day 90. All physicochemical analysis was done in triplicate.
Bacterial community structure and diversity
In the bioinformatics and statistical analyses, 26 736 reads were captured, with an average of 6684 counts per sample. After filtering to a minimum of 4 counts and a prevalence of 20%, the data set contained 513 ASVs, which were further reduced to 292 final ASVs. The relative abundance of taxa correlates directly to the observed ASVs, although it may not represent the actual biomass changes.37,36 The dominant bacterial phyla observed across all samples were Acidobacteria, Actinobacteria, Firmicutes and Proteobacteria, as shown in Figure 2A. Hydrocarbon contamination in the HPS-1 caused a decrease in the abundance of Proteobacteria and Firmicutes compared to the UPS. Acidobacteria and Actinobacteria increased in response to the pollution. In the mixed soil sample with chicken droppings at t = 1 day (MAS-1), Firmicutes showed a threefold increase compared to HPS-1 and a 4-fold reduction compared to MAS-90 (end of remediation).

(A) Bacteria phyla of microbial communities. (B) Genera of microbial communities.
However, Acidobacteria and Actinobacteria significantly increased in MAS-90 relative to MAS-1. The mixed sample without nutrients at t = 1 day (MSS-1) had the highest percentage of Proteobacteria ASVs compared to the other samples, which all had Proteobacteria as the most abundant phylum. The MAS-90 and MSS-90 showed a high abundance of Actinobacteria, but it was highest in MSS-90. Phyla with less than 1% abundance (labelled ‘others’) appeared in UPS-1 (with the lowest frequency), MSS-90 and HPS-1 (with the highest frequency). The phyla comprising the 1% include Bacteroidetes, Planctomycetes, Deinococcus-Thermus and Chloroflexi, as depicted in the differential abundance heatmap in Supplementary Figure S.1.
The first 50 genera in Figure 2B show bacteria in the unpolluted and polluted samples, although most are not known as hydrocarbon degraders. However, genera with Methyl- prefixes are well-represented in the microbiome. The 10 dominant ASVs were aligned with Caballeronia, Bacillus, Paenibacillus, Paraburkholderia, Novosphingobium, Cohnella, Methylocystis, Pseudolysinimonas, Massilia and Alloiococcus. Rare taxa ASVs in UPS-1 observed in the polluted samples include Fontibacillus, Gulosibacter, Methyloferula, Conexibacter, Altererythrobacter, Acidocella, Methylocystis, Novosphingobium, Paraburkholderia, Oceanobacillus, Bradyrhizobium, Pseudolysinimonas, Cohnella and Caballeronia. Caballeronia had the least abundance (0.3%) in UPS-1, the highest in HPS-1, and mid values in other soil samples with varying hydrocarbon pollution levels. Bacillus decreased from 32.65% in UPS-1 to 7.32% in HPS-1. Paenibacillus abundance dropped from 13.59% in UPS-1 to 3.26% in HPS-1, with the highest value in the nutrient-treated sample (MAS-1, 21.08%) and around 10% in the samples without nutrient treatment (MSS-1 and MSS-90). The Massilia genus had the highest abundance in MAS-1, followed by UPS-1, but was least abundant in MSS-1 and MSS-90. The percentage abundance of Methylocystis in the samples (UPS-1, HPS-1, MSS-90, MAS-90, MSS-1 and MAS-1) showed a descending order. The dominance of Paraburkholderia, Alloiococcus, Cohnella, Novosphingobium and Pseudolysinimonas did not follow a clear pattern. However, Cohnella, Alloiococcus and Pseudolysinimonas had the highest abundance in MSS-90, with 12.81%, 11.37% and 8.74%, respectively. Novosphingobium had an abundance of 11.31% in MAS-1, while Paraburkholderia had 11.42% in MAS-90.
The ASV alpha diversity measure in Figure 3 suggests that the treated samples exhibit the highest alpha diversity. This is supported by indices dependent on richness, such as Observed, Chao1 and ACE, which show a sequential decline in diversity across the samples MAS-1, MAS-90, MSS-1, HPS-1, UPS-1 and MSS-90, as illustrated in Supplementary Figure S.3. Considering both relative abundance and richness, the Shannon, Simpson and inverse-Simpson indices rank the MAS-90, HPS-1 and MAS-1 samples as the top 3, in terms of diversity. The discrepancies in these results may be because the Simpson index is less affected by rare taxa, while the Shannon index is more robust against variations in library size. To delve deeper into the diversity distinctions between the control and treated samples, beta diversity was assessed using the permutational multivariate analysis of variance (PERMANOVA) test. The findings were depicted on a principal coordinate analysis (PCoA) plot (refer to Supplementary Figure S.2) generated using the Bray-Curtis distance. The first 2 principal components account for 26.1% and 32.3% of the observed variance.

Alpha diversity measures of microbial communities under control and treatment conditions.
Functional prediction analysis
Functional pathway analysis based on the 16S rRNA genes revealed a diverse range of pathways, as shown in Supplementary Figure S.4. The enzyme commission (EC) numbers and KEGG Orthology (KO) associated with the predicted pathways (PWs) underwent the DAA. The results indicated no significant difference between the control and treated samples, as depicted in Supplementary Figure S.5. Supplementary Figure S.4 illustrates several hydrocarbon metabolism pathways, including aromatic compound degradation, catechol degradation, protocatechuate degradation, the Calvin-Benson-Bassham (CBB) cycle and methanol oxidation. Among all the soil samples, the CBB pathway showed the highest expression. The protocatechuate pathway exhibited the least expression, as indicated in Figure 4.

Predicted functional potential of microbial communities based on PICRUSt2 analysis, showing the proportion of sequences associated with selected metabolic pathways.
Figure 5 presents a plot of selected hydrocarbon-degrading enzymes against sequence proportion. Among the 3 hydrocarbon-degrading enzymes analysed (alkane-1-monooxygenase, 3-carboxyethylcatechol-2,3-dioxygenase, 2,4-dichlorophenol-6-monooxygenase), alkane-1-monooxygenase showed the highest expression across the samples, and its expression was found to be statistically significant.

Predicted functional potential of microbial communities based on PICRUSt2 analysis, showing the proportion of sequences associated with selected metabolic pathways.
Discussion
Hydrocarbon pollution is a pervasive issue in the Niger Delta, resulting from petroleum exploration, transportation activities and artisanal operations. This pollution not only degrades the land but also harms beneficial microbiota, eukaryotic organisms and human health. Among various remediation approaches, bioremediation is prioritised for its sustainability benefits. In this study, we employed microbial remediation to decontaminate heavily polluted farmland affected by artisanal refining activities. To monitor the remediation progress, we used infrared spectrometric techniques to track the concentration of TPHs over time. At the same time, metagenomics was applied to observe the dynamics of hydrocarbon-degrading bacteria and their functions. After the 90-day remediation period, we observed the expression of enzymes related to hydrocarbon metabolism pathways and a mix of classical and non-classical hydrocarbon-degrading bacterial genera. Moreover, a significant reduction in the initial TPH concentration was achieved, indicating the effectiveness of the remediation process.
Polluted soil differs from unpolluted soil in its physicochemical and microbial properties, often requiring remediation to restore its health. Hydrocarbon contamination induces significant physicochemical alterations in soil, affecting its pH stability, ionic exchange capacity and organic matter composition. The presence of petroleum-based compounds triggers acid-base fluctuations, predominantly reducing pH, 38 although in some cases, it may lead to an increase. 39 The pH trend observed in this study indicates a progressive shift towards increased soil acidity. Hydrocarbon pollution disrupts microbial proliferation and enzymatic functions crucial for soil homeostasis. Essential macro-elements such as nitrogen (N), phosphorus (P) and organic carbon (C) deplete due to biochemical stressors and impaired nutrient cycling. Our results reveal a significant depletion of nitrogen (N) and phosphorus (P) as was found in the study of Zhang et al. 40 Hydrocarbon compounds also chelate metallic ions, modifying their solubility and bioavailability, which increase toxicity levels and reduce nutrient assimilation by plants. In affected soils, vegetation diversity is severely diminished, as observed during site clearing for field bioremediation, with only toxicity-resistant species or those suitable for phytoremediation remaining. Singh et al 41 recognised ryegrass and tall fescue as highly effective in remediating xenobiotic hydrocarbon contaminants. Structurally, contamination compacts soil matrices, increasing bulk density while diminishing hydraulic conductivity and aeration potential, thereby obstructing root penetration and water movement. Hydrocarbon-induced hydrophobicity impairs moisture retention, leading to erratic water distribution and restricted microbial colonisation. Furthermore, microbial ecosystems undergo selective pressure, with hydrocarbon-degrading taxa, while more sensitive microbial populations decline. These combined alterations culminate in soil infertility and ecosystem destabilisation, necessitating remediation interventions such as chemical oxidation, adsorptive amendments or bioremediation to restore equilibrium and reinstate soil functionality. One key benchmark for initiating remediation is the predetermination of the concentration of TPHs, which varies based on environmental regulations and soil type. Remediation is typically necessitated when TPH concentrations surpass 100 ppm in residential and agricultural soils or 500 ppm 42 in industrial environments, as these thresholds indicate potential ecological and human health risks. Effective bioremediation strategies include microbial degradation, phytoremediation and soil amendments to enhance natural recovery processes. The strategy employed in this study was chicken dropping, which is effective in reducing contaminant levels, improving soil structure and restoring microbial diversity.
In rural settings, chicken wastes are often collected in open dumps, posing significant health risks. Unmanaged waste can harbour pathogens and emit harmful gases, such as ammonia and methane, which can lead to respiratory issues, infections and contamination of soil and groundwater through leachates resulting from nutrient overload. This contamination affects agricultural produce and human health through the food chain 43 Robust waste management practices are essential to mitigate these hazards, yet such measures are frequently beyond the reach of economically disadvantaged communities. Effective management involves the systematic collection and treatment of poultry litter through aerobic composting, often enhanced by co-composting with other organic materials, and the use of covered storage to limit pathogen proliferation, odour and leachate contamination while transforming the waste into a nutrient-rich soil amendment. 44 Although small-scale operations may manage chicken manure with relative ease, increased poultry production demands more sophisticated and scalable waste processing systems. In addition, capturing biogas for energy use is becoming an increasingly common strategy. Economically, repurposing chicken waste for remediation and agricultural applications reduces reliance on synthetic fertilisers, improves crop yields, lowers remediation costs and creates new revenue streams and job opportunities in the waste processing sector. While there are initial costs associated with proper handling, processing and application, a thorough cost-benefit analysis alongside site-specific risk assessments can ensure that the long-term economic and ecological benefits outweigh these challenges. The utilisation of poultry waste in bioremediation presents a tangible advantage, facilitating the efficient attenuation of soil contaminants through microbial and physicochemical interactions.
The significant reduction of TPH, observed in this study, can be attributed to a combination of physical, chemical, environmental and biological factors. Key factors that influence TPH attenuation include dilution (physical factor), the nature and properties of pollutants (chemical factors), pH, temperature, nutrients and moisture (environmental factors), the initial microbial community, enzymes and adaptability (biological factors). The optimal ranges of these factors were confirmed at the same study site by Obieze et al. 45 Various biological factors contributed to the overall attenuation of TPH through biodegradation, such as in situ biosurfactant production, cometabolic effects from vertical and horizontal microbiota, bacterial surface phenomena, 46 the abundance of adapted biocatalysts, horizontal functional gene transfer and supply of limited nutrients. Specifically, in this study, the application of poultry manure contributed a 0.3% to the reduction in TPH concentration. Previously, Okpashi et al 47 recorded a percentage efficiency of 0.06%. Several scholars have affirmed the efficacy of chicken droppings in soil TPH attenuation.48 -50 The effectiveness of chicken droppings in hydrocarbon remediation is partly attributed to their content of micro- and macro-nutrients. The addition of chicken droppings, which contain total nitrogen (0.9%–2.3%) and available phosphorus (0.5%–1.5%),51,52 stimulated the microbial community and primed biological factors that enhanced biodegradation, although its effect was not statistically significant with the bioattenuation regime. The statistical evaluation conducted on 4 experimental groups and 2 control groups yielded 15 P-values. Among these, 5 exhibited false negatives (type II errors) and 1 false positive (type I error). After adjustment with the Bonferroni correction, the type I error was corrected. Entry 2 in Supplementary Table S.1 exhibits a P-value of .115854189, reflecting a greater mean difference of 99.99% compared to Entry 4, which demonstrates a statistically significant P-value of .000000007941 despite a relatively lower mean difference of 99.96%. This underscores the stochastic nature of statistical inference, indicating that sole reliance on P-values for significance determination may not always be robust or reliable. Type II errors have been documented in disciplines such as mathematics, biostatistics and epidemiology, as noted in a correspondence published in Nature by Kafi and Ansari-Lari. 53 The authors formally asserted that a 10% variance between the comparative pairs should yield statistical significance. However, all instances of non-significance in the TPH concentration exhibited a deviation exceeding 12%.
The residual contaminant concentrations in both nutrient-amended and unamended treatments remain above the threshold values prescribed for effective site remediation. This indicates that the 3-month remediation period is inadequate to achieve complete bioremediation of the impacted site. In Nigeria, regulatory oversight of hydrocarbon-contaminated site remediation is primarily governed by the DPR, now integrated into the Nigerian Upstream Petroleum Regulatory Commission (NUPRC). The EGASPIN serves as the principal framework for environmental compliance in the sector. 54 Under EGASPIN, a target value of 50 mg/kg is prescribed for TPHs in soil, representing the acceptable concentration below which environmental and public health risks are considered negligible. 55 This target is founded on risk-based environmental management principles aimed at safeguarding human health, ecological receptors and groundwater resources. This concentration is considered sufficiently low to minimise long-term toxicological effects from chronic exposure, particularly in residential and agricultural settings. It reflects a precautionary threshold, aligned with international best practices from jurisdictions of other countries, where similarly conservative values are adopted for sensitive land uses. Moreover, this benchmark provides a pragmatic endpoint for remediation efforts, offering regulators and stakeholders a scientifically grounded criterion for determining site closure, reuse or continued monitoring while balancing environmental integrity with socio-economic feasibility in the Nigerian context. Conversely, an intervention value of 5000 mg/kg is delineated, signifying the concentration at which immediate remediation is mandated due to the potential for significant ecological and human health hazards. 56 These benchmarks are essential in delineating contamination severity, guiding risk-based corrective measures and informing the design and implementation of remediation strategies tailored to site-specific conditions. Contemporary remediation strategies increasingly advocate for bioremediation as a sustainable and cost-effective approach to managing hydrocarbon-contaminated sites. In certain applications, the process involves the amendment of soil with organic nutrient sources, including poultry manure such as chicken droppings
Poultry manure, primarily composed of the faeces and urine of poultry birds, is a globally significant organic waste, with chicken producing the largest quantity. It contains up to 13 essential plant nutrients, including zinc, iron, manganese, molybdenum, boron, copper and cobalt, as well as major nutrients like nitrogen, phosphorus, potassium, calcium and magnesium, which are crucial for microbial stimulation and growth. 57 Fresh poultry waste typically comprises 1.8% potash, 2.5% phosphate, 3% nitrogen and 40% water, although these values can vary depending on the type of bird, feed source, feed ration, medication and the ratio of droppings to litter. 58 In its fresh state, poultry manure poses significant environmental and health risks as mentioned above. Therefore, it must undergo composting to reduce the community of pathogenic microorganisms, during which the water content and nutrient levels decrease. Composted poultry manure generally contains around 1.2% nitrogen, 0.4% phosphorus and 0.05% potassium, which are available for uptake by plants and microorganisms. 59 Besides providing essential nutrients, poultry droppings enhance the soil’s physical properties by improving its granular structure. Additionally, poultry manure is rich in heterotrophic bacteria, including some that degrade hydrocarbons. Amending polluted soil with poultry droppings has been shown to promote hydrocarbon degradation, achieving 60% to 85% degradation within a 24-month treatment period. 51 ,60 -63 Thus, incorporating poultry waste into soil achieves several benefits: it enriches the soil’s nutrients, improves its physical properties, repurposes waste, and enhances bioremediation through both biostimulation and bioaugmentation.
The dominance of Proteobacteria across all samples underscores its essential role in the soil biogeochemical cycling of nitrogen, sulphur and carbon. 7 However, the decline in Proteobacteria abundance in HPS compared to the pristine soil sample indicates the toxic impact of hydrocarbons on sensitive members of this phylum. This trend has been observed in several studies on hydrocarbon-induced bacterial diversity, including that of Duran and Cravo-Laureau 64 . Interestingly, the reduction in Proteobacteria is compensated by an increase in Actinobacteria and Actinobacteria phylotypes in unpolluted soil (UPS-1), suggesting that these phyla harbour significant populations of hydrocarbon-degrading bacteria, as demonstrated by Kim et al, 65 Behera and Das 66 and Ren et al. 67 However, Iturbe-Espinoza et al 68 reported a contrasting scenario, where hydrocarbon pollution caused a decreased relative abundance of Actinobacteria and Acidobacteria. The dynamics of Actinobacteria across the samples indicated a positive response to hydrocarbons but an inhibition at high concentrations, with the highest abundance observed in the 90-day remediated soil samples. This study suggests that certain genera of Acidobacteria thrive in high hydrocarbon concentration environments, while certain Actinobacteria members prefer moderate concentrations. The shift in microbial composition also indicates that chicken droppings significantly enriched the soil with Firmicutes, a finding consistent with previous research.69,70 However, during the bioremediation, the abundance of Firmicutes decreased markedly, implying the presence of hydrocarbon-sensitive Firmicutes phylotypes. The minor phyla distribution, which is highest in the HPS, suggests an affinity for hydrocarbon degradation. Among these phyla, Bacteroidetes, Planctomycetes, Deinococcus, Chloroflexi, Gemmatimonadetes and candidate division WPS-2 are represented in Supplementary Figure S.1. These minor phyla have been previously documented in hydrocarbon-contaminated soil and associated degradation processes.71,72
The study area’s predominant bacterial genera featured classical hydrocarbon degraders, including Bacillus, Paenibacillus, Paraburkholderia, Methylocystis, Methyloferula, Acidocella, Bradyrhizobium, Cohnella and Paraburkholderia. Conversely, rare-rich taxa that also play a role in hydrocarbon degradation include Fontibacillus, Gulosibacter, Methyloceanibacter, Conexibacter, Altererythrobacter, Caballeronia and Pseudolysinimonas. These emerging hydrocarbon-degrading bacteria likely originate from the soil and the introduced nutrient source (chicken droppings). This finding aligns with previous research demonstrating the presence of hydrocarbon-degrading bacteria in both soil and chicken manure.73,74 In addition, the significant reduction in bacterial phyla in the HPS confirms the inhibitory effect of hydrocarbons on many bacterial species. However, certain resistant bacteria with potential hydrocarbon-degrading capabilities emerged from the Bacteroidetes and Planctomycetes phyla in hydrocarbon-saturated soil samples. Similar trends of increased Bacteroidetes and Planctomycetes in response to hydrocarbon pollution have been reported by Jiao et al 75 and Sazykina et al, 71 although Sun et al 76 demonstrated a contrary result. These findings suggest that microbial community structure is influenced by the specific types of available pollutants and the representative genera within each phylum. For example, the increased presence of Caballeronia, Paraburkholderia and Fontibacillus in the heavily and mildly polluted soil samples serves as biomarkers for hydrocarbon pollution. Conversely, the dominance of Micropepsis and Lysinibacillus in UPS may indicate a high sensitivity to hydrocarbon contamination. The former category can be referred to as hydrocarbonoclastic bacteria. While Paraburkholderia is a well-known hydrocarbon-degrading bacterium, Caballeronia and Fontibacillus are not hydrocarbonoclastic bacteria, particularly in the context of the Niger Delta terrestrial ecosystem.
Genera not initially recognised as known biodegraders may develop hydrocarbon-degrading abilities or resistance through prolonged exposure to hydrocarbons, a process known as acclimatisation. The evolving presence and significance of certain genera, such as Altererythrobacter (Proteobacteria), Caballeronia (Proteobacteria), Fontibacillus (Firmicutes), Gulosibacter (Actinobacteria) and Conexibacter (Actinobacteria), in hydrocarbon pollution remediation studies warrant investigation. Although Altererythrobacter is less well-known compared to Paraburkholderia, Bacillus, Paenibacillus, Methylocystis, Methyloferula, Acidocella, Bradyrhizobium and Cohnella (identified in this study), it is recognised for its hydrocarbon-degrading properties. Wu et al 77 reported Altererythrobacter as a co-dominant genus in hydrocarbon-polluted soil in Shaanxi Province, China. In addition, Cai et al 78 demonstrated that Altererythrobacter spp. can degrade hexadecane from soil. This genus has also been implicated in effective polycyclic aromatic hydrocarbon (PAH) degradation in soil. 79 Huang et al 80 Identified Altererythrobacter among PAH-degrading bacteria in marine environments. Caballeronia emerged as the second most abundant genus in the unamended soil during a rhizoremediation study by Lee et al. 81 A genome-driven biodegradation screening in Japan confirmed that Caballeronia sp. The 19CS4-2 efficiently degrades 3-chlorobenzoate within 24 hours. 82 Lopez-Echartea et al 83 also observed the dominance of Caballeronia alongside Burkholderia and Paraburkholderia in a phytoremediation study in Finland. Interestingly, the Caballeronia-Burkholderia-Paraburkholderia (BCP) complex is featured in this work. Conexibacter (another less-recognised genus) was identified as a predominant taxon in crude oil-polluted tropical soil in Nigeria. 84 It was also dominant in a co-contaminant risk impact assessment in aged Australian soil. 85 Hung et al 86 established that Conexibacter is an efficient PAH-degrading bacterium. Gulosibacter molinativorax was identified in a culture-dependent approach as a hydrocarbon-degrading bacterium derived from polluted wastewater. 87 Yang et al 88 also detected Gulosibacter in a hydrocarbon-polluted soil metagenome through the polymerase chain reaction – denaturing gradient gel electrophoresis (PCR-DGGE). These findings highlight that Altererythrobacter, Caballeronia, Conexibacter and Gulosibacter are under-reported hydrocarbon-degrading bacteria in the Niger Delta region, which is known for its rich microbial diversity. Thus, the Niger Delta terrestrial ecosystem harbours a variety of hydrocarbon-degrading bacteria, including genera not previously documented.
Fontibacillus is a potentially emerging hydrocarbon-degrading genus, although no published evidence currently supports its role in hydrocarbon degradation. A novel strain isolated from a warm spring exhibits 93.6% to 96% sequence similarity to Paenibacillus, a known hydrocarbon-degrading genus. 89 Fontibacillus is a less-studied genus of spore-forming bacteria that shares similarities with Paenibacillus but has distinct ecological roles, although comparative studies remain limited. 90 This genus was notably abundant in the heavily contaminated soil, with reduced frequency in the mildly polluted soil, suggesting a potential role in hydrocarbon degradation. This could be the first report linking Fontibacillus to hydrocarbon degradation in soil in the Niger Delta soil. These findings merit further genomic investigation, as using genus-level shifts to assess microbial responses to hydrocarbon pollution is typically more suited for aquatic environments.91,92 Tracking of higher taxa (such as phylum) in soil environments may provide a more accurate understanding of bacterial responses to hydrocarbon perturbation, as indicated by this study (Guerra et al, 93 and Gao et al 94 ). Responses of bacteria to hydrocarbons are usually reflected in metabolism.
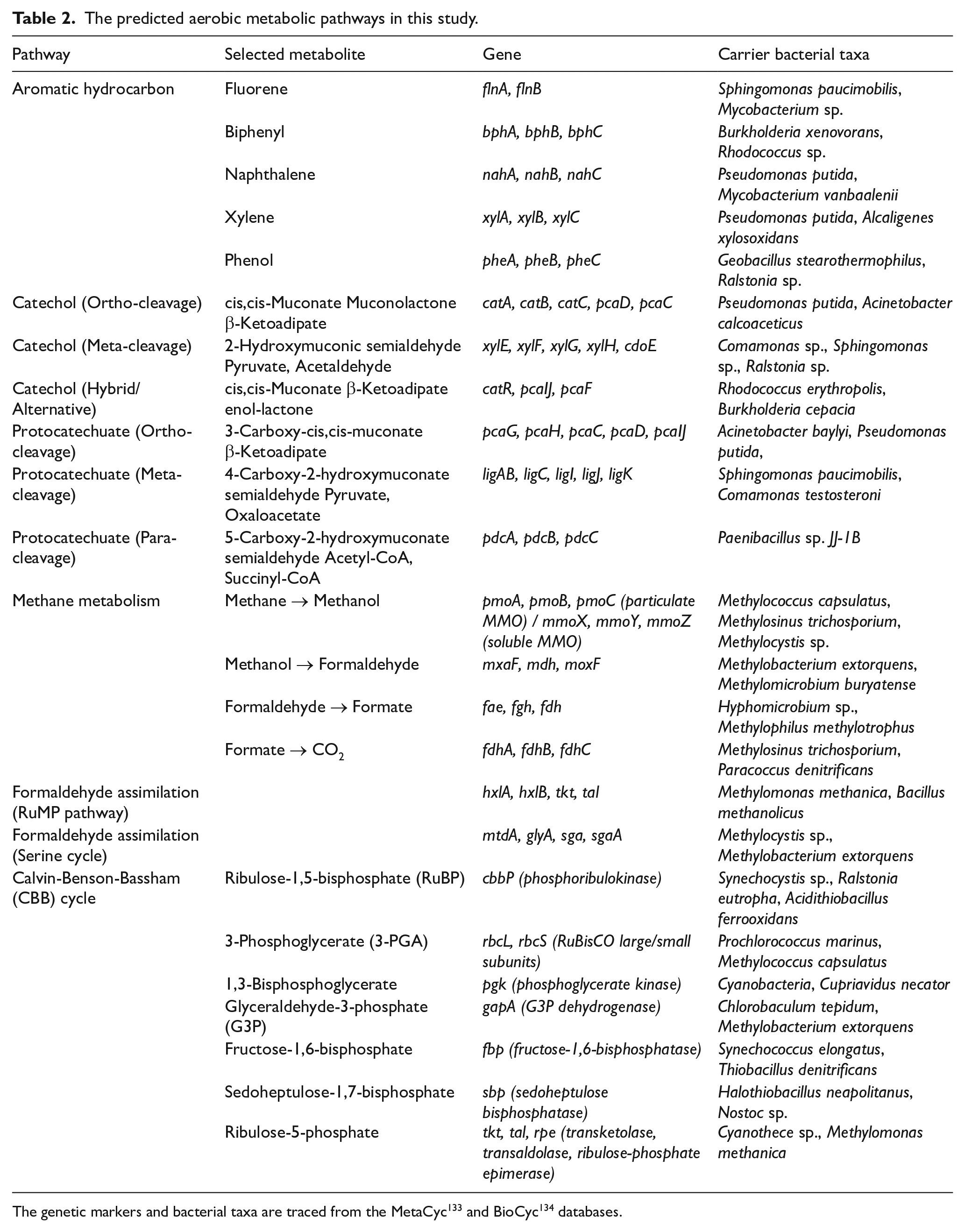
During the bioremediation process, several metabolic pathways were predicted, including methane metabolism, alkane metabolism and degradation of halogenated aromatic compounds. The catabolic pathways of these compounds are prevalent in environments contaminated with hydrocarbons. The prediction of chemo-markers like catechol and protocatechuate, derived from salicylates and phthalates, respectively, provides strong evidence of aromatic hydrocarbon degradation. 95 The predicted occurrence of pyruvic acid and acetaldehyde, alongside succinic acid and acetyl-CoA, respectively, indicates the metabolism of protocatechuic acid and catechol via ortho- and meta-cleavage pathways, as was demonstrated by Zhao et al 96 and Nghiem. 97 Acetyl-CoA is a metabolite of alkane degradation, driven by alkane 1-monooxygenase in the beta-oxidation pathway. Both ortho- and meta-cleavage metabolites enter the central metabolic (tricarboxylic acid (TCA) cycle) pathway, with carbon dioxide marking mineralisation. Formate from methanol, derived from methane, also enters the TCA cycle and produces carbon dioxide, which is used in the Calvin-Benson-Bassham (CCB) cycle for carbon fixation. 98 This interconnected pathway, shown in Figure 6, predicts a high presence of hydrocarbon-degrading bacteria, methanotrophs and autotrophic bacteria in the study site. These pathways are dominantly influenced by aerobic respiration. Aerobic degradation plays a crucial role by activating aromatic hydrocarbons with the insertion of molecular oxygen, forming dihydroxy intermediates. This process is catalysed by enzymes known as dioxygenases. The dihydroxy intermediates are further degraded into smaller compounds that enter the central metabolic pathway. Polycyclic aromatic hydrocarbons and BTEX (benzene, toluene, ethylbenzene and xylene) are commonly targeted for biodegradation by bacteria and fungi in polluted environments. The 1-ring protocatechuate and catechol pathways observed in this study predict efficient microbial biodegradation of aromatic compounds, as these pathways are closely linked to the central metabolic pathway responsible for compound mineralisation. Wu et al 95 demonstrated that catechol is associated with BTEX biodegradation and is linked to functional genes contributed by synergistic bacterial partners. Catechol intermediate molecules are involved in the metabolism of various compounds, including naphthalene and anthracene, 99 chrysene, 100 phenanthrene and fluorene 101 as well as pyrene. 102

Proposed summary pathways existing in the study site using the predicted metabolites, enzymes and cycle derived from Supplementary Figure S.4 (pathways), Figure 4 (metabolites and pathways) and Figure 5 (enzymes).
Conversely, protocatechuate has been reported as a biomarker for naphthalene degradation by Rhodococcus ruber OA1 103 phenanthrene oxidation by Micrococcus strain, 104 fluorene utilisation by Sphingobacterium sp. KM-02, 105 pyrene by Pseudomonas aeruginosa PFL-P1 106 and benzo(a)pyrene biodegradation by Serratia marcescens S217. 107 These aromatic hydrocarbons’ degradations are directed by key functional genes like PAH-RHDαβ (alpha and beta sub-units of PAH-ring hydroxylating dioxygenase gene), nahAC (naphthalene dioxygenase genes), phnAc (phenanthrene dioxygenase gene) and bphA (biphenyl dioxygenase gene), which encode key enzymes like protocatechuate-3,4-dioxygenase, catechol-2,3-dioxygenase and catechol-1,2-dioxygenase.108 -111 These enzymes break down aromatic compounds. Several other specific enzymes are expected to drive pathways identified in Figure 4. The presence of these enzymes predicts active biodegradation of n-alkanes and aromatic hydrocarbons during the remediation process at the polluted site. It is important to note that aromatic hydrocarbons are generally more persistent and resistant to biodegradation than n-alkanes, which are catalysed by alkane-1-monooxygenase, the most expressed enzyme in this study. Alkane monooxygenase plays a vital role in catalysing the degradation of medium-chain alkanes (n-C8 to n-C18), which include diesel, kerosene and gasoline fractions of aliphatic hydrocarbons that constitute a major part of crude oil. The gene encoding alkane monooxygenase, such as alkB and its variants like alkW and alkM, is commonly found in hydrocarbon-degrading bacteria across diverse ecosystems, including terrestrial, aquatic and specialised environments. In addition, this gene is highly prone to horizontal gene transfer. The alkB gene facilitates alkane degradation through terminal and sub-terminal pathways, producing primary and secondary alcohols. Dioxygenase enzymes also contribute to the bi-terminal and Finnerty pathways, utilising double-oxidised alkanes as substrates. 112
The presence of alkB genes in hydrocarbon-degrading bacteria enhances the degradation of various hydrocarbon fractions, including PAHs, BTEX, isoalkanes, short-chain alkanes and even alkanes longer than C20. 113 Notable long-chain alkane degraders possessing alkane monooxygenase include Dietzia species, Acinetobacter pitti sw-1, Acinetobacter sp. M-1, Geobacillus thermodenitrificans NG80-2, Pseudomonas aeruginosa DN1 and Alcanivorax borkumensis SK2.114 -116 Excellent short-chain alkane degraders like Mycobacterium sp. NBB4, Amycolicicoccus subflavus DQS3-9A1T and Rhodococcus sp. NRRL B-16531 also possess alkane monooxygenase.117 -119 The AlkB protein exerts a synergistic effect due to its roles in signal transduction, activation of the C-H bond and global regulation.120 -123 Although alkane-1-monooxygenase is more specific to medium-chain alkanes, it also shows a closer affinity to short-chain alkanes compared to long-chain alkanes. 124 In this study, the predicted biomarker pathways for methane metabolism identified were the CCB cycle and methanol oxidation.
Methane metabolism is governed by two genetically distinct variants of methane monooxygenase (MMO): the di-iron-containing soluble methane monooxygenase (sMMO) and the copper-dependent, membrane-associated particulate methane monooxygenase (pMMO), both regulated by functional genes. Additional insights into functional genes linked to hydrocarbon degradation are presented in Table 2. Methanotrophs, the bacteria that metabolise methane, primarily rely on pMMO, although some specialised groups, such as Verrucomicrobiota methanotrophs, use lanthanides in the transformation of methanol. 125 Methane oxidation occurs via 3 main pathways: the RuMP (ribulose monophosphate) cycle, used by type I methanotrophs (Gammaproteobacteria); the Serine cycle, used by type II methanotrophs (Alphaproteobacteria); and the CCB cycle, used by Verrucomicrobiota methanotrophs.112,126 The Verrucomicrobiota methanotrophs utilise methane as an energy source to fix CO2, highlighting their potential to mitigate methane emissions and sequester CO2. This suggests that Verrucomicrobiota methanotrophs play a crucial role in sustainably addressing the 2 most prevalent greenhouse gases, contributing to environmental sustainability and climate change mitigation. These methanotrophs utilise lanthanide-dependent MMO to convert methane into methanol, which is then further metabolised into formaldehyde and other intermediate metabolites for assimilation. Members of the Verrucomicrobiota phylum include *Methylacidiphilum and *Methylacidimicrobium, 127 while Proteobacteria genera include Methylvimicrobium, 128 *Methylomonas, *Methylocella, *Methylocapsa, *Methylocystis, 129 Methylococcus, 130 Methyloceanibacter, Methylocystis, Methylosinus 131 and *Methyloferula. 132 The asterisked genera are acidophilic methanotrophs. Notably, Methyloferula, Methylobacterium and Methylocystis were among the first 50 dominant genera identified in this study. The presence of Methyloferula suggests that the study site is rich in methanotrophs with sMMO, capable of degrading a wide range of short-chain hydrocarbons. In polluted soil metagenomes, a consortium of fungi, bacteria and algae often promotes cometabolic and synergistic effects for hydrocarbon degradation. The phylotypes observed in the results indicate that novel bacteria are active players in this complex and polluted soil, contributing to the biodegradation and attenuation of hydrocarbons.
The predicted aerobic metabolic pathways in this study.
The residual concentrations of TPHs in both amended and unamended soils indicate that degradation exceeded 90%, with no significant difference, despite the amended soil showing a higher percentage of TPH degradation. Although not inconceivable, this unexpected outcome may be attributed to an excessive nutrient supply, the bio-inhibiting impact of hydrocarbon pollution, the possible inertia induced by high molecular weight hydrocarbons and the activated nature of the contaminated site. The unamended soil achieved its success through enhanced natural attenuation, suggesting that the mildly polluted state of the impacted site stimulated the microbiome to degrade hydrocarbons effectively. This confirms that the site already harbours competent hydrocarbon-degrading microbes, classifying it as activated soil, soil with self-remediating capability. 135 Activated soil typically contains enriched microbial communities, nutrients and organic matter that facilitate petroleum hydrocarbon breakdown. The presence of diverse hydrocarbon-degrading bacteria and fungi accelerates contaminant decomposition via enzymatic processes. 136 Barbeau et al 137 used activated soil to achieve a 99% reduction of pentachlorophenol in 130 days. Such activated soil, by way of enhancement, promotes oxygen availability, which is crucial for aerobic degradation pathways, and improves soil structure, promoting better water retention and nutrient distribution to support microbial metabolism. 138 Research indicates that microbial activators in soil significantly boost enzymatic activity, leading to higher degradation rates of hydrocarbons, including complex compounds such as PAHs. 138 Optimising activated soil composition with targeted microbial strains and nutrients further enhances its efficiency in remediating hydrocarbon-contaminated environments. However, it is necessary to understand the preliminary characteristics to develop a guide for the optimisation. For instance, a self-remediating soil site should exhibit a microbial cell count exceeding 103 CFU, a near-neutral pH, an optimal C:N:P ratio of 100:10:1 and a soil porosity of 30% to 40%.139 -141 The addition of composted chicken waste shifts the bioremediation approach from natural attenuation to a combination of bioaugmentation and biostimulation. However, incorporating a 25% composition of chicken waste into unconfirmed activated soil, as is the case in this study, results in an excess nutrient supply.
Excess nutrients can disrupt soil hydrocarbon degradation by encouraging fast-growing non-degrading microbes that prioritise available nutrients over hydrocarbons, reducing degradation efficiency. 142 Previous studies have shown that biostimulation does not significantly outperform natural attenuation. In alignment with the findings of this study, Koshlaf and Ball 143 proposed that this effect is attributable to a metabolic shift within the microbial community, wherein alternative carbon and nutrient substrates provided by the amendment are preferentially utilised over diesel oil degradation. This shift raises additional inquiries into how microbial metabolic pathways are modulated by environmental amendments and the potential impact on remediation efficiencies. The increased microbial respiration driven by fast-growing, non-degrading populations can induce oxygen depletion. This oxygen deficit subsequently impedes aerobic degradation processes by establishing a microaerobic environment that is suboptimal for hydrocarbon-degrading microorganisms. Moreover, the degradation kinetics of hydrocarbons by microaerophilic or anaerobic bacteria are considerably lower compared to those exhibited by aerobic microbes. Moreover, chemical binding of nutrients to soil particles or organic matter sequesters essential components for microbial hydrocarbon metabolism, restricting their uptake and slowing both manure decomposition and hydrocarbon metabolism. 144 Manure can also bind heavy metals, reducing their toxicity; however, this process restricts microbial interactions and enzymatic functions necessary for hydrocarbon degradation. This bioavailability bottleneck creates a feedback loop that prolongs contamination persistence. Again, if manure fails to supply nutrients in the proper ratios, it may worsen imbalances and further impede hydrocarbon degradation and microbial recovery, especially in polluted soils.
Hydrocarbon-contaminated soils present a multifaceted challenge to microbial communities due to the simultaneous presence of diverse toxicants such as heavy metals and petroleum hydrocarbons. These contaminants exert selective pressure that drastically reduces microbial diversity by inhibiting sensitive taxa and impairing key enzymatic and metabolic functions. For instance, heavy metals can bind to critical proteins and enzymes, altering their tertiary structures and obstructing the functionality of metabolic pathways essential for cellular energy production and detoxification. Concurrently, petroleum hydrocarbons may induce oxidative stress and disrupt membrane integrity, further impeding microbial growth and activity. 145 When manure is introduced into such an environment with the intention of bioaugmenting the microbial population, its effectiveness is compromised by the pre-existing hostile conditions. The microorganisms present in manure, although diverse, are generally not preadapted to the elevated concentrations of toxicants, and they often fail to establish themselves in competition with indigenous, stress-tolerant species. 146 These native microbes have typically undergone selective adaptations, such as the development of specialised efflux systems or enzymatic detoxification pathways, which confer a competitive advantage under contaminant exposure. Assuming these conditions prevail in this study, the introduction of exogenous microorganisms will likely intensify interspecific competition rather than enhancing bioremediation, consequently reducing their effective contribution to hydrocarbon attenuation.
Assuming that the chicken waste exerts its anticipated beneficial effects, the amended soil will initially exhibit a markedly enhanced rate of hydrocarbon degradation. This acceleration persists until a secondary inhibitory phase, hereafter referred to as the second inertia effect, manifests following the substantial degradation of low-molecular-weight hydrocarbons, at which point high-molecular-weight hydrocarbons, characterised by lower bioavailability and heightened recalcitrance, become predominant. The first inertial is the period of acclimatisation of the exogenous microbes. Meanwhile, the unamended soil continues its intrinsic degradation process, ultimately resulting in TPH concentrations that converge with those of the amended soil despite the latter’s initial performance advantage. Over prolonged remediation periods, degradation can potentially resume provided that the recalcitrance of the residual hydrocarbons does not exceed the degradative capacity of the exogenously introduced microbial community. This phenomenon has been exemplified by Oghoje et al, 147 where a 20% chicken waste amendment achieved its optimal degradation performance at day 168 in HPS. As the structural complexity of the remaining hydrocarbons increasingly overwhelms the catalytic capabilities of microbial enzymes, further degradation is progressively hindered by factors such as insolubility and sequestration onto soil particles, a binding effect that, while protecting the compounds from essential oxidative and reductive processes, simultaneously limits their biotransformation. Consequently, this phase in the bioremediation process often necessitates auxiliary interventions, including the deployment of biosurfactants and/or bioaugmentation with specialised microbial consortia or genetically adapted strains, to enhance the breakdown of these persistent compounds.
Conclusions
This study demonstrated field-scale bioremediation of HPS using a straightforward biopile technique, in which chicken droppings were utilised as a nutrient source to stimulate both indigenous and introduced microbiomes. Over the 90-day remediation period, spectrometric, metagenomic and bioinformatics analyses confirmed (and predicted the presence of hydrocarbon-affiliated enzymes and pathways) the presence of both conventional hydrocarbon-degrading bacteria and novel bacterial genera that may have contributed to the degradation of methane, n-alkanes and aromatic compounds in the petroleum-contaminated soil. The minimal variation in residual TPH concentrations between the amended and unamended soils suggests a lack of statistically significant differences, notwithstanding the marginally superior performance of the amended treatment. This outcome may be ascribed to factors such as the site’s intrinsic natural attenuation capacity, potential inhibitory interactions between the contaminated soil matrix and the introduced poultry manure, nutrient oversaturation or the inherently reactive and variable chemical characteristics of hydrocarbon pollutants. Furthermore, the residual TPH levels post-bioremediation imply that a 90-day remediation period may be inadequate for achieving effective soil decontamination.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322251371117 – Supplemental material for Metagenomic Insights Into Biopile Remediation of Petroleum-Contaminated Soil Using Chicken Droppings in Rivers State, Nigeria
Supplemental material, sj-docx-1-bbi-10.1177_11779322251371117 for Metagenomic Insights Into Biopile Remediation of Petroleum-Contaminated Soil Using Chicken Droppings in Rivers State, Nigeria by Emmanuel O Fenibo, Rosina Nkuna and Tonderayi Matambo in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
We thank the University of South Africa for granting us access to their laboratory facilities. Our sincere thanks go to Dr Ramganesh S. for providing invaluable support in data analysis and to Dr Ijoma G. for her expert guidance during the laboratory sessions. Their contributions were instrumental in ensuring the successful completion of this project.
Author Contributions
Emmanuel O. Fenibo played a major role in conceiving the study, curating the data, conducting laboratory experiments and contributing to manuscript writing. Rosina Nkuna was responsible for statistical analysis, figure preparation and overall methodological development. Tonderayi Matambo provided essential supervision throughout the research process and manuscript revision. All authors have thoroughly reviewed and approved the final manuscript for submission.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Data availability statement
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.