
Abstract
Millions of individuals suffer from chronic infections caused by bacterial biofilms, resulting in significant loss of life. Pseudomonas aeruginosa stands out as a major culprit in causing such chronic infections, largely due to its antibiotic resistance. This pathogen poses a considerable threat in healthcare settings, particularly to critically ill and immunocompromised patients. The persistence of chronic and recurrent bacterial infections is often attributed to bacterial biofilms. Therefore, there is an urgent need to discover novel small molecules capable of efficiently eliminating biofilms independent of bacterial growth. In this project, an in silico drug discovery approach was employed to identify nine halogenated-phenazine compounds as allosteric inhibitors of the LasR protein. The LasR is a key transcription factor that triggers other quorum-sensing systems and plays a crucial role in biofilm formation and activation of virulence genes. By inhibiting LasR, specifically targeting its allosteric site, the dimerization of LasR and subsequent biofilm formation could be prevented. Molecular docking and simulations, coupled with binding energy calculations, identified five compounds with potential as anti-biofilm agents. These compounds exhibited higher binding affinities to the distal site, suggesting their structural capability to interact with allosteric site residues of the LasR protein. Based on these findings, it is proposed that these compounds could serve as promising leads for the treatment of biofilm and quorum-sensing-related infections.
Keywords
Introduction
Most of the persistent infections stem from bacterial biofilms, commonly characterized by their pathogenic properties. Data provided by the National Institutes of Health (NIH) suggests that biofilm formation is associated with 65% and 80% of microbial and chronic diseases, respectively.1-3 According to Ali et al in 2023, bacterial biofilms typically resist the human immune system and drugs. In essence, the efforts of some existing antimicrobial agents are rendered futile. 4 One of the most important ways to control and eradicate diseases linked to bacterial biofilm is to understand biofilm formation in bacteria. 2 Most of the research on bacterial pathogenesis focuses on acute infection; however, the chronic form of infection is less attended to.5,6 Nevertheless, for decades, it has been evident that most chronic bacterial infections result from bacteria’s capacity to flourish within aggregates encased in biofilms. Bacteria residing in biofilms are highly shielded from antibiotics, antimicrobial agents, and host defenses, making them exceedingly challenging or often nearly impossible to eliminate. 7 An illustrative instance of biofilm participation in chronic infections is observed with Pseudomonas aeruginosa in the lungs of individuals afflicted by cystic fibrosis (CF).8-11 In light of the bacterial organization in biofilm, this chronic infection is incurable and ultimately leads to the demise of individuals with CF.10,11
Bacterial biofilms predominantly contribute to chronic and recurrent bacterial infections. Consequently, there is an urgent necessity to identify novel small molecules that function independently of bacterial growth to efficiently eliminate biofilms. Among the bacterial pathogens, P. aeruginosa is an opportunistic human pathogen that can cause severe long-term infections in people with weakened immune systems. 12 The infamous persistence of this pathogen in clinical settings is ascribed to its capacity to generate biofilms resistant to antibiotics. 12 There are reports that patients with persistent infections, such as chronic lung infections, persistent wound infections, and persistent rhinosinusitis, may develop highly structured biofilms.13,14 There are reports that over 90% of chronic wound infections are thought to be significantly influenced by biofilms, which impair wound healing. Compared to bacterial infections alone, patients who have bacterial infections in conjunction with other conditions have a fourfold increased risk of dying.15,16
According to Ali et al, 4 Zhang et al, 6 Bano et al, 7 and Bowler et al, 17 a biofilm is an intricate assemblage of bacteria that sticks to both biotic and abiotic surfaces. Biofilm is encased in an extracellular mucus matrix composed of proteins, lipids, and polysaccharides. Antibiotic bioavailability is hampered by biofilms, which can also lower antibiotic levels within the biofilm and result in drug resistance.5,13,17 P. aeruginosa and other biofilm-forming bacteria exhibit a 1000-fold increase in resistance to antibiotics when biofilm is formed.7,18 Therefore, innovative approaches to the eradication of biofilms are desperately needed. Quorum sensing (QS) is essential for P. aeruginosa biofilm development and the control of virulence factors that provide the bacteria resistance to antibiotics. In QS, diffusible chemical signaling molecules called autoinducers facilitate cell-to-cell contact among different types of bacteria. Bacterial population growth leads to an accumulation of signaling molecules in the surrounding environment, which promotes gene expression, biofilm development, and population density regulation. Critical roles in virulence, biofilm formation, and tolerance to antibiotics, detergents, and chemicals are played by two interconnected QS systems.6,19 The transcriptional activator lasR and the acyl-homoserine lactone synthase lasI make up the first system (Las). An acyl-homoserine lactone synthase (rhlI) and a transcriptional activator (rhlR) comprise the second system (Rhl).6,19 The LasR is the main transcriptor that triggers the other QS systems, this receptor plays a crucial role in biofilm formation, and activates virulence genes. In contrast, the las system is at the top of the QS hierarchy. N-(3-oxododecanoyl) homoserine lactone (3-O-C12-HSL) is produced by the autoinducer synthase LasI. This leads to the activation of virulence genes such as lasB, lasA, apr, and toxA by the lasR-encoded transcriptional activator LasR. LasR’s architecture consists of two domains: an N-terminal DNA binding domain (DBD) and a C-terminal ligand-binding domain (LBD). Findings from Bottomley et al 8 convey that the LasR’s autoinducer (3-O–C12–HSL) binds to the receptor, which results in stability and then goes through dimerization. The target DNA promoter is subsequently attached to by the resulting LasR–autoinducer homodimer complex, which stimulates the transcription of genes. According to Vetrivel et al, 20 inhibiting LasR may interfere with QS and prevent the production of biofilms. Recently, research confirmed this idea by observing that P. aeruginosa isolates with mutant LasR genes were not biofilm makers. 21 This highlights the importance of LasR as a possible target for controlling the formation of biofilms by inhibiting QS.
P. aeruginosa strains can produce 90%–95% of the blue phenazine pigment known as pyocyanin. 22 This finding highlights that P. aeruginosa uses pyocyanin as a defense against other bacteria to prevent biofilm perturbation. With the excellent antibacterial properties of pyocyanin, Huigens and coworkers took a special interest in microbial competition strategy and investigated the potential of phenazine antibiotic-inspired compounds to eradicate persistent bacterial biofilms in Staphylococcus aureus. 23 From their findings, a series of halogenated phenazines (HPs) that potently eradicate bacterial biofilms in S. aureus were identified, and future work aims to translate these preliminary findings into ground-breaking clinical advances for the treatment of persistent biofilm infections. Based on the findings by Huigens and colleagues, 23 this study aims to explore the potential antibiofilm activity of these compounds against P. aeruginosa LasR protein and hence provide a molecular basis to explain their mechanism using a computational approach.
In this investigation, the primary binding site of these compounds was identified through the virtual screening technique. Findings from the virtual screening indicate that all nine compounds bind to an allosteric site. Molecular dynamics (MD) simulations demonstrated the stability of five compounds bound at the allosteric site, which could potentially hinder LasR dimerization and thereby inhibit biofilm formation. Given these outcomes, we suggest that these compounds could represent promising alternative candidates for addressing infections related to biofilm and QS.
Methods
Protein selection and preparation
The three-dimensional (3D) structure of the LasR protein of P. aeruginosa in complex with the anti-activator and autoinducer; PDB ID: 6V7X was obtained from the Protein Data Bank (https://www.rcsb.org). Chain A was chosen and prepared by the removal of water molecules, heteroatoms, and the anti-activator (Aqs1) from the protein structure. 24 Histidine residues were protonated, and partial charges were added to the protein structure to ensure charge balance on protein structures. The backbone structures of the protein were energy minimized using AMBERff4SB, and Gasteiger charges were computed using Antechamber in chimera. We followed the methods of Mensah et al.
Ligand preparation and structural optimization
Ligand modeling was performed using Spartan ’14 (Wavefunction Inc., Irvine, California, USA), and structural optimizations and energy minimization were performed using the density functional theory B3LYP/6-31G* basis set. 25 Compounds were saved in .pdb and .sdf file format for further analysis. Figure 1 shows the compounds considered for the study.
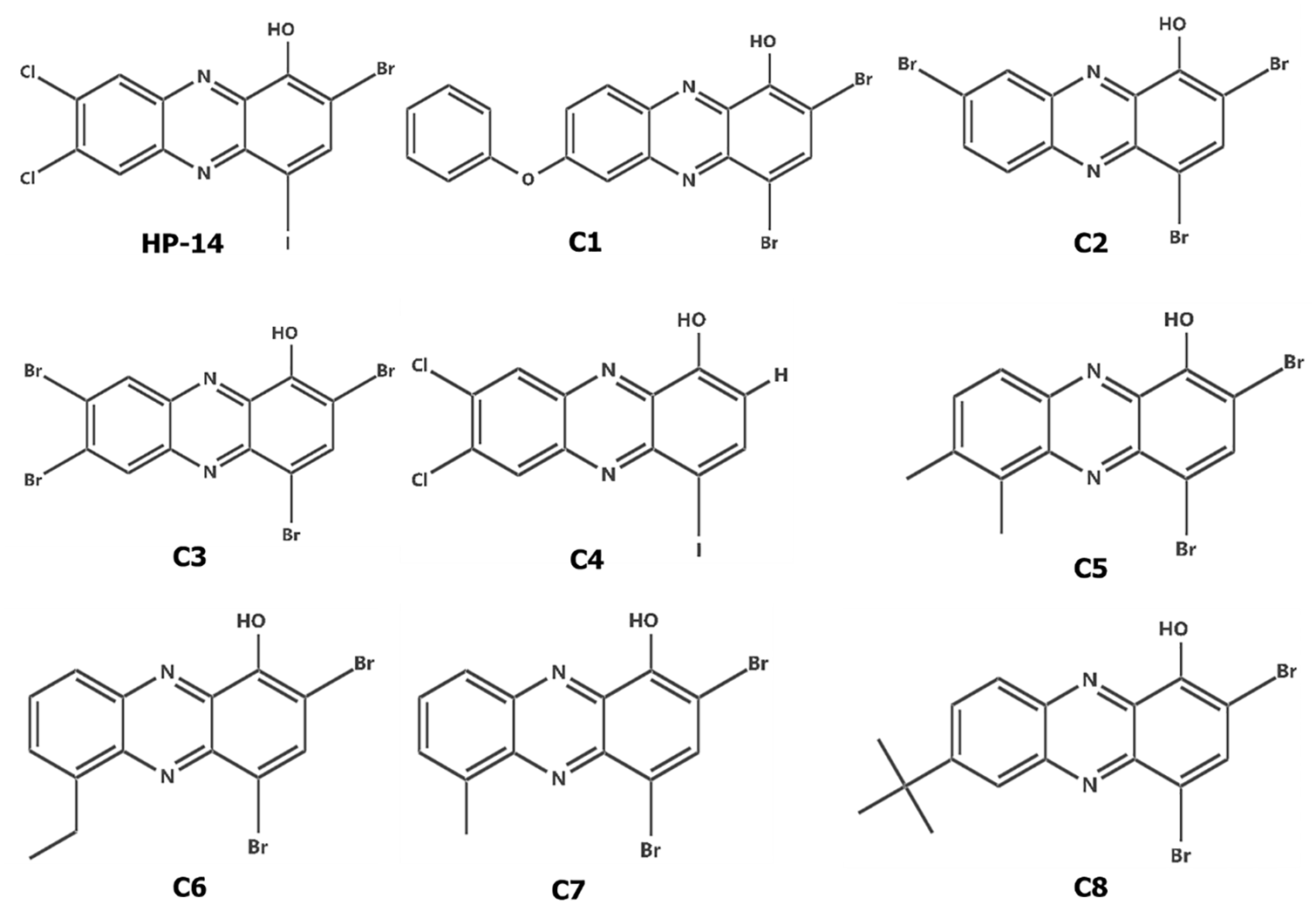
Halogenated phenazine compounds considered for this study.
Molecular docking
Virtual screening
The virtual screening was performed with PyRx v0.824.26,27 The methodology proceedings followed that of Kwarteng et al. We employed the AutoDock Vina28 Lamarckian Genetic algorithm and Empirical Free Energy Scoring function within the PyRx v0.8 interface. Grid dimensions used in the study were set to cover the entire protein with dimensions (Å): X = 50.9317; Y = 44.2851 and Z = 57.7354 with center: X = 22.7209; Y = 4.0930 and Z = −13.2669. All prepared ligands were targeted against the LasR protein in a blind docking manner. A total of nine different poses were generated for each ligand, and the lowest energy poses were considered.
MD simulation study
The protein-ligand complexes were set up for an all-atom MD simulation for 200 ns using GROMACS V.2018.6 28 on the Lengau cluster (Centre for High-Performance Computing, Cape Town). The 3D crystal structures of proteins were saved in a .pdb format. Ligands were prepared by subjecting CHARMM force field and hydrogen addition and saved in a .mol2 format. Using the CGenFF force field server, 29 ligand topologies were attained and saved in a .str format, while protein topologies were generated with the pdb2gmx tool with the CHARMM36 force field. The complexes were placed in a dodecahedron simulation box, and a TIP3P water model was added for solvation to be attained. By the addition of Na+ and Cl– ions, ionization of the systems was achieved. The systems were subjected to energy minimization using the steepest descent method to reduce steric clashes for 50,000 steps. Temperature and pressure were kept constant at some point in the simulation process. With this, the systems were subjected to equilibration under normal volume and temperature (NVT) and normal pressure and temperature (NPT) conditions, ensuring constant numbers of particles, volume, pressure, and temperature ensembles. This process was conducted at temperatures up to 300 K and pressures up to 1 bar over a duration of 100 ps. To account for electrostatic and van der Waals interactions, the computation of long-range electrostatic interactions utilized the Particle Mesh Ewald (PME) method, and cut-off distances for both Coulombic and van der Waals interactions were set at 1.2 nm. Time steps of 2 fs were defined for the production run, with coordinate trajectories saved every 10 ps. Three-dimensional periodic boundary conditions (PBC) were implemented, succeeded by a 200 ns MD production run.
Post-MD analysis of 200 ns simulated systems was done to check for the stability of the ligands and other critical analyses for the receptors used in the study. Before conducting any analysis, all output trajectory files were corrected using the gmx trjconv command in GROMACS. For stability analysis, values for the radius of gyration (Rg), root mean square deviation (RMSD), and root mean square fluctuation (RMSF) were generated, saved in a .xvg format using gmx gyrate, gmx rms and gmx rmsf programs, respectively. The generated .xvg files were then plotted using the XMGRACE tool. Eigenvalues and eigenvectors were generated using gmx covar and gmx anaeig, respectively. By using generated files of both eigenval.xvg and eigenvec.xvg files, the principal component analysis (PCA) of the complexes were evaluated and plotted using the XMGRACE tool.24,30-32 We followed the methods of Kwarteng et al.
MM-PBSA binding energy calculations
To evaluate the energetic contributions necessary for ligand stability, the Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) approach 33 was implemented to calculate the binding energies of the ligands when bound to the receptor. The MM-PBSA approach evaluated the binding energy by analyzing energetic components linked to alterations in potential energy within a vacuum environment. These components encompassed bond angle and torsional energies, van der Waals and electrostatic interactions, as well as the de-solvation process involving various species. De-solvation was assessed for polar and nonpolar solvation energies utilizing an implicit solvation model alongside the configurational entropy linked to complex formation. The estimated binding energies incorporated molecular mechanical potential energy (EMM), as well as parameters for polar (Gpol) and apolar (Gapol) solvation. The binding energy (Ebinding) of the system is estimated as

where Ecomplex is the total free energy of the target–ligand complex, Etarget and Eligand are the total free energies of the individual target (receptor) and ligand in a solvent, respectively.
The individual binding free energy of each component is expressed as

where Emm represents the molecular mechanics energy terms, Gsolv represents the solvation energy terms. It is noteworthy that the entropic factor (ΔTS) was excluded from the computation, primarily due to its considerable computational complexity. Moreover, there are reports suggesting that the net effect of the entropic component is frequently insignificant. 28 This explains why the binding energy is designated as Ebinding instead of ΔG. The Emm is made up of all bonded and non-bonded energies in the system, thus, can be expressed as

where Ebonded is the bonded interactions consisting of bond, angle, dihedral, and improper interactions. Enonbonded represents the non-bonded interactions that include both electrostatic (Eelec) and van der Waals (EvdW) interactions, which are calculated using Coulomb and Lennard–Jones potential functions, respectively. The solvation energy term (Gsolv) is expressed as

where Gpolar represents polar solvation energies and Gnonpolar is the nonpolar solvation energies. Gpolar, which is the electrostatic contribution, is calculated by solving the Poisson–Boltzmann equation. The non-electrostatic term of solvation energy, Gnonpolar, includes repulsive and attractive forces between solute and solvent generated by cavity formation and van der Waals interactions, respectively. We followed the methods of Alexander Kwarteng et al.
Results
High-throughput virtual screening
Results from the high-throughput virtual screening predicted the binding site of the ligands used for the study. The ligands were binding at the distal site of the LasR protein (see Figure 2) using the PyRx software. PyRx generated nine different poses of docked ligands with varied RMSD. Poses with the lowest binding score and corresponding minimum RMSD were considered for further analysis. We considered all ligands because their poses were at the distal site and reported their lowest binding scores as given in Table 1.

Binding poses of halogenated-phenazine compounds from virtual screening with AHL (yellow).
Docking scores of compounds from the high-throughput virtual screening.

Abbreviations: HP, halogenated-phenazine.
MD simulations
MD simulations were performed in explicit solvent to explore the allosteric potential of the compounds used for the study. As the P. aeruginosa LasR protein performs its signal transductions with the aid of an autoinducer, N-3-oxo-dodecanoyl-L-Homoserine lactone (3-oxo-C12-HSL, also known as acyl-homoserine lactone [AHL]), complexes, with AHL bound at the active site and HPs at the distal sites, were used to provide intrinsic insights during the simulation period. An unbound protein (apo form) was used to explore the protein dynamics in the bound and unbound states. Post-MD analysis of the ternary complexes was investigated. The PCA of the ternary complexes was evaluated considering the whole protein structure as well as the DNA-binding domain. Comparative studies were done on all complexes to better understand their dynamics throughout the simulation period.
Protein–AHL–HPs (ternary) complexes
Stability analysis
As part of stability investigations, several pieces of information were evaluated, including the RMSD of the ligands, RMSF of protein side chains, Rg, and the PCA of the protein. By taking into consideration these characteristics and accounting for dynamic aspects that are not captured by molecular docking alone, the MD simulations provide an in-depth insight into the stability of ligand-protein interactions as previously described.24,31
Ligands analysis
RMSD of the ligands
The RMSD obtained from the MD trajectory serves as a vital measure in evaluating the protein-ligand complex. By selecting various atoms for comparison, RMSD examines the coordinates of two superimposed structures. The RMSD is a fundamental technique for predicting the conformational stability of both the ligand and protein.
From the RMSD of the ligands, the extent of deviation of the ligands was observed. Five compounds from the studied compounds were bound at the distal site of the LasR protein with an RMSD value of less than 2 nm.31,34-36 These compounds include C2, C3, C4, C6, and HP-14. For the analysis of compound C2, an average RMSD value of 1.5 nm was observed consistently throughout the simulation period (see Figure 3A, represented by the green trajectory). This indicates that C2 remained highly stable and formed strong interactions with amino acid residues at the distal site. Similarly, for compound C3, an average RMSD value of 0.75 nm was observed (see Figure 3A, represented by the blue trajectory), indicating strong interactions with amino acid residues and overall stability. Detailed examination of compound C4 revealed that it remained bound at the distal site, although a deviation was observed between 48 and 65 ns due to flipping of the compound (see Figure 3A, represented by the yellow trajectory). Despite this flipping, C4 maintained stability at the distal site, with an average RMSD value of 1.75 nm. Compound C6 also remained stable at the distal site throughout the simulation, with slight flipping observed during the simulation period (see Figure 3A, represented by the gray trajectory). The average RMSD value of C6 was 0.95 nm. Similarly, compound HP-14 exhibited stability at the distal site throughout the simulation, resembling the observations made for C2, C3, C4, and C6 (see Figure 3A, represented by the magenta trajectory).
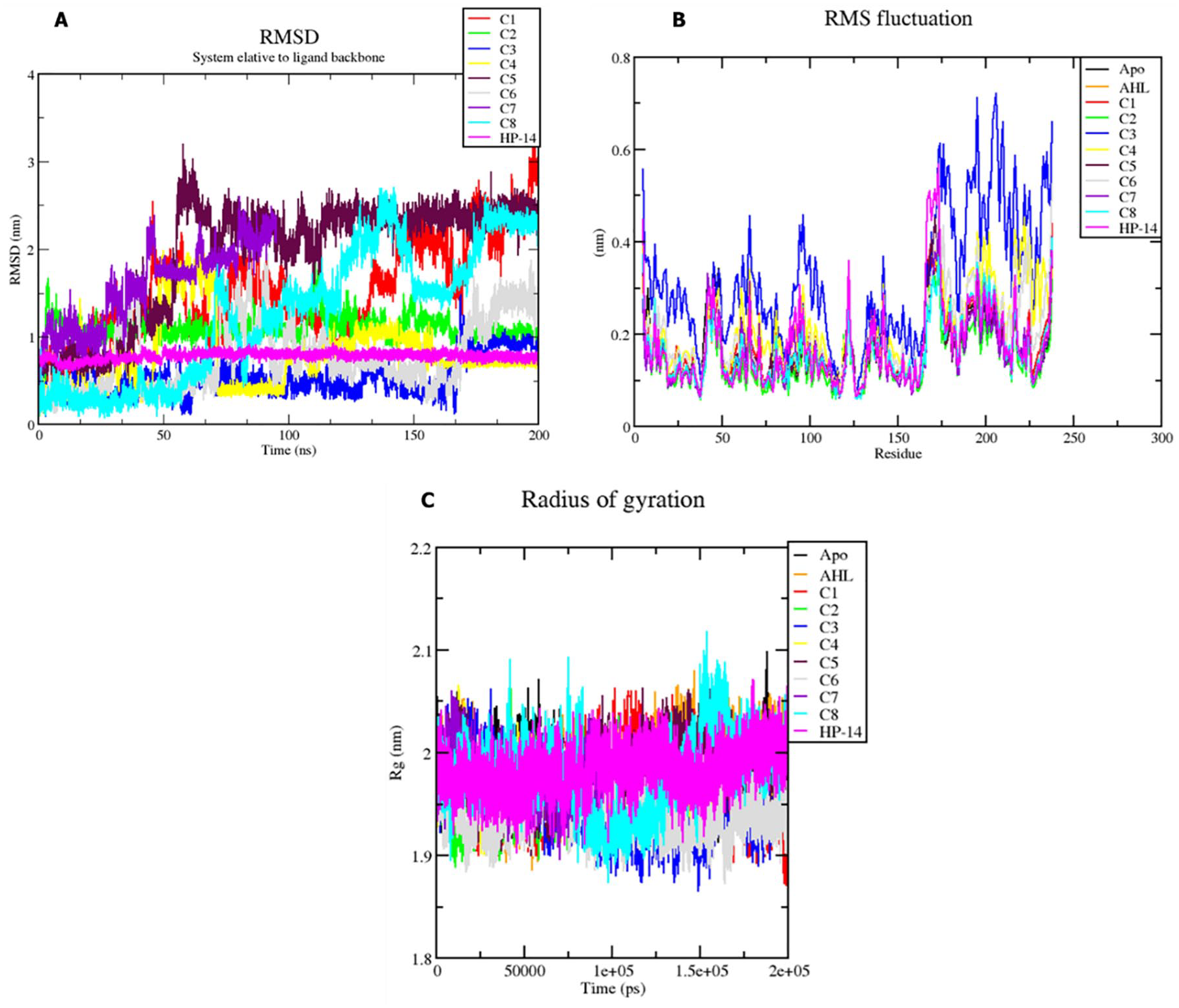
Trajectory analysis of protein-ligand complexes: (A) RMSD of ligands, (B) RMSF of side chain fluctuations in bound and unbound proteins, and (C) radius of gyration of bound and unbound proteins.
In contrast, four compounds, C1, C5, C7, and C8, departed from the distal site during the 200 ns simulation. These compounds lost interactions with the amino acids at the distal site, rendering them unstable (see Figure 3A). Detailed analysis showed that C1 initially bound to the distal site but experienced major deviations from 0 to 170 ns, leading to its transition into the bulk solvent from 180 to 200 ns. Compound C5 initially exhibited stability from 0 to 50 ns but deviated due to binding at a different site away from the distal site, specifically at the DNA-binding domain of the protein (see Figure 3A). Compound C7 remained bound at the distal site from 0 to 60 ns but eventually vacated to the bulky solvent due to the loss of strong interactions with amino acids. Compound C8 was initially bound at the distal site from 0 to 125 ns but later bound at different sites within the DNA-binding domain of the protein from 130 to 200 ns (see Figure 3A). We followed the methods of Alexander Kwarteng et al.
Protein analysis
RMSF of protein side chains
RMSF indicates the residual mobility of amino acids.24,34 A substantial degree of mobility and instability in the receptor is indicated by the high RMSF values of amino acid residues. Conversely, stable and rigid receptors are defined by amino acid residues with low RMSF values. The overall RMSF of the amino acid residues was calculated for binary complex (lasR–AHL) as well as the ternary complexes. Analysis of the unstable ligands shows that the unstable ligands such as C1, C5, C7, and C8 had a minimal influence on the side chain of the amino acid residues. This comparison was evident when the RMSF of the unstable ligand complexes was compared to the apo and the AHL–bound complexes. When the unstable ligands vacate the distal site, the protein assumes a stable structural conformation that recognizes the presence of the AHL. This explains that the native ligand had less impact on the structural feature of the protein. Hence, the protein in the unbound state had a similar structural integrity as when the native ligand was bound to it and when the unstable ligands exited the distal site (see Figure 3B). This observation is confirmed as the native ligand is a natural substrate of the protein, and hence, when bound at the active site, signal transduction is induced, and the structural integrity of the protein is maintained. Considering the stable ligands, a similar scenario exists for the unbound ligands. Significant fluctuations of the residues were observed in the C2, C4, C6, and HP-14 at the ligand and DNA-binding domains (see Figure 3B). For the C3 complex, higher fluctuations were observed in the protein. Significant fluctuations were noted in the DNA-binding domain (DNA-BD) as well as the LBD (see Figure 3B-blue). We followed the methods of Alexander Kwarteng et al.
Rg of the protein backbone
The compactness, stability, and folding of the protein structures are indicated by Rg.31,34-36 In this study, the computation of Rg was based on both binary and ternary complexes intrinsic dynamics throughout the simulation. Observations made were that no drastic unfolding was observed for the apo-form of protein and the AHL-bound (binary) complex as witnessed from the Rg. Considering the ternary complexes, slight compactness was noted for unstable ligands (C1, C5, C7, and C8) ternary complexes. A similar case was observed for stable ligand (HP-14) ternary complex, but for C2, C3, C4, and C6 ternary complexes, there was an increase in the compactness of the protein. As Rg is the distribution of atoms of proteins around its axis, 26 this implies that the stable ligands bound at the distal site caused an increase in the compactness of the protein by restricting and causing rampant motions of residues at the ligand-binding and DNA-binding domains. Furthermore, the compactness of the C3–ternary complex was the highest as compared to the others (see Figure 3C-blue trajectory).
Principal component analysis
Overall protein backbone and DNA-binding domain
The PCA was utilized to examine the collective motion of protein atoms within the ternary complexes, providing insights into the overall stability of these complexes. By applying the MD trajectory to a phase space, a set of eigenvalues was generated, aiding in elucidating the protein’s flexibility. Furthermore, an analysis of overall flexibility was conducted using the trace of the diagonalized covariance matrix. Higher trace values indicate increased model flexibility, which is consistent with previous RMSF investigations. Tertiary conformations along eigenvectors 1 and 2 were represented in a scatter plot obtained by sampling proteins and their complexes in phase space using a projection of the C-alpha atom. As illustrated in Figure 4, both the binary (lasR–AHL) complex and the apo-protein occupied a relatively larger region in the phase space. Conversely, the C3 ternary complex exhibited a larger occupancy zone, similar to unstable ligands and several stable ligands, indicating relatively higher flexibility in the C3 ternary complex.
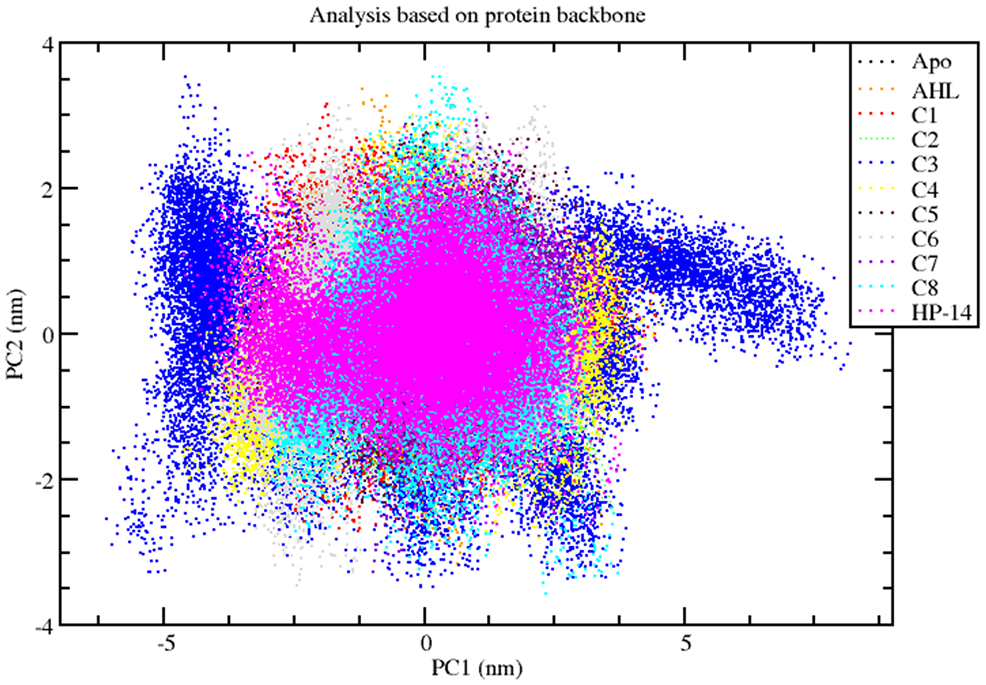
Two-dimensional projection motions for all the systems as principal component analysis.
Hence, a further PCA of the DNA-binding domain of the protein was evaluated. This is because the monomeric form of 3-oxo-C12 HSL, a natural AHL, stabilizes and dimerizes to two LasR subunits on binding to LasR. Following ligand interaction, the resulting homodimer gains the ability to bind DNA via the DNA-binding domain, hence initiating transcriptional modifications. 37 Findings from this analysis suggest that the C3 compounds caused an increase in the flexibility of the domain when compared to AHL (see Figure 5B; Figure 6; C6-blue and AHL-black). This is also backed by the RMSF analysis (see Figure 3B).

Two-dimensional projection motions of the DNA-binding domain as principal component analysis: (A) C2, (B) C3, (C) C4, (D) C6, and (E) HP-14.
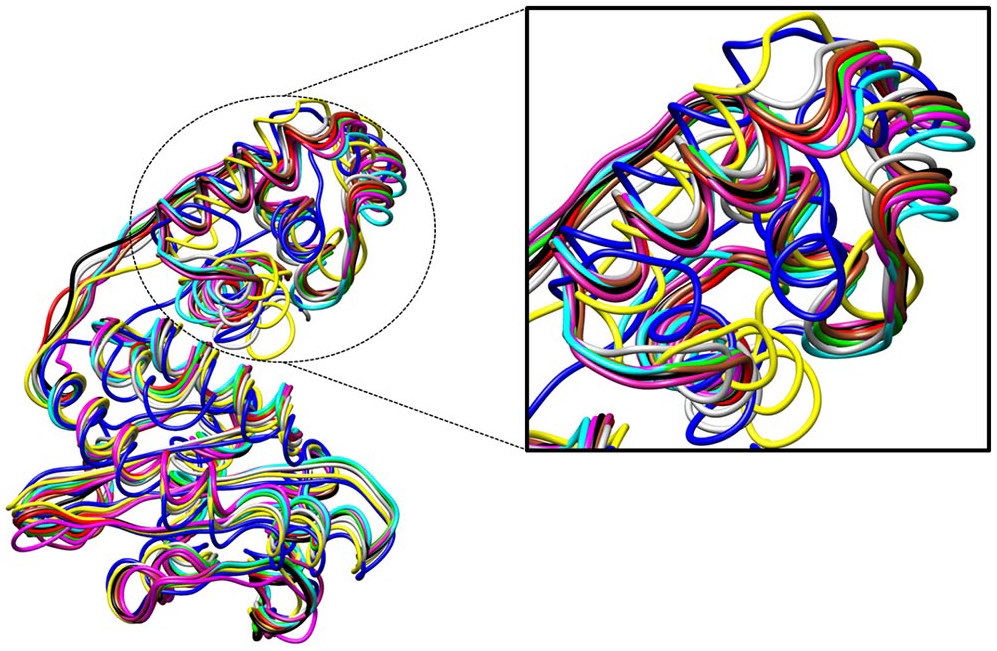
Average structure of all systems that shows the structural deviation of the protein throughout the simulation.
Free energy landscape analysis
The first two principal components, PC1 and PC2, are used as reaction coordinates, using GROMACS inbuilt scripts (g_covar, g_anaeig, and g_sham). Free energy landscape (FEL) analysis was performed to look at the unique binding conformation. Examining the FEL plot indicates that higher energy states are indicated by red to yellow regions, while dark blue to violet labels reflect the energy minima and energetically favored structural conformation. 38 The data unambiguously demonstrate that ligand binding altered the free energy within the global free energy minimum. In addition, each structure exhibited distinct global energy minima, signifying stable states. This suggests that the receptor-ligand ternary complexes adopt a resilient and stable structure (depicted in Figure 6). Snapshots of ligand-protein ternary complexes coinciding with global energy minima were obtained from the MD trajectory, illustrating the preferred conformations of LasR (as depicted in Figure 6).
MM-PBSA binding free energy calculations
Although molecular docking offers the lowest energy and the ligand-bound protein’s binding conformation, it does not take into account the protein’s natural conformational changes, solvation and ionic effects, or the entropic contributions to the total binding free energies.31,34-36 Molecular dynamics simulations are utilized to estimate the stability and corresponding energy of ligands bound to their targets, taking into account docking restrictions. The calculated binding free energies were utilized to evaluate the compounds’ affinities for the allosteric pocket residues of LasR. Various energy components, such as van der Waals, electrostatic, polar, and nonpolar solvation energies, contribute to the overall binding energy. Analyses of the complexes revealed significant contributions from van der Waals, electrostatic, and nonpolar energies to the binding process. Conversely, polar solvation energy negatively impacted the binding efficiency of all complexes. The outcomes of the MM-PBSA calculations are summarized in Table 2, focusing on the binding energies of C2, C3, C4, C6, and HP-14, which exhibited stability at the allosteric site throughout the simulation duration.
The table represents the van der Waals, electrostatic, polar solvation, SASA, and binding energy in kJ/mol for predicted hit compounds.

Discussion
The QS plays a crucial role in P. aeruginosa, facilitating the formation of biofilms and the regulation of virulence factors, which contribute to the bacteria’s resilience against antimicrobial agents. Apart from biofilm production and resistance to detergents, antibiotics, and biocides, two interconnected QS regulatory mechanisms are pivotal for virulence. 24 The first system, Las, comprises the transcriptional activator lasR and the AHL synthase lasI. The second system, Rhl, consists of a transcriptional activator (rhlR) and an AHL synthase (rhlI). 24 Signaling molecules produced by the Las and Rhl QS systems are detected by their respective LuxR-type receptors. In addition, there are two other QS systems: the Integrated Quorum System (IQS) and the Pseudomonas Quinolone Signal (PQS), which utilize quinolone as the signaling molecule. 39 Among the machinery in P. aeruginosa QS systems, the Las system initiates gene expression in the QS hierarchy. The LasR, a transcriptional regulator, plays a promising role in the pathogenesis of P. aeruginosa by mediating the expression of QS genes. The pathogenicity of P. aeruginosa can be controlled by inhibiting the LasR protein, which holds the topmost position in the hierarchical QS network.37,40 The LasR is stabilized and undergoes dimerization when its autoinducer binds to it.24,37,40
The native LasR signaling molecule N-(3-oxo-dodecanoyl)-L-homoserine lactone when present at a quorate concentration binds to LasR and activates the QS pathway. 37 Bottomley et al, demonstrated that the binding of the autoinducer results in ligand stabilization and protein folding via van der Waals’ interactions within a hydrophobic core and via H-bond linking of distal elements of secondary structure. The hypothesis from this study is that the AHL 1-oxo group links Tyr-56 in the β 2–α3 loop to Ser-129 of strand β3, and this is concomitant with protein folding. 37 Vetrivel et al., 37 demonstrated that the polar head group of the autoinducer forms hydrogen bonds with the amino acid residues Tyr56, Trp60, Asp73, and Ser 129 while the nonpolar tail group binds with hydrophobic residues, including Leu36, Leu40, Ille52, Val76, and Leu125 at the ligand binding pocket of LasR. These findings suggest that the AHL-dependent plays a major role in the folding switch of LasR and is a probable representation of QS regulation in all LuxR homologs. Again, these interactions make the autoinducer shield the ligand-binding pocket from bulk solvents and sequester the hydrophobic residues near the active site pocket into a favorable hydrophobic environment.24,37 Concomitant with protein folding, LasR dimerization occurs, and the stabilization of the LasR in its active homodimer state binds to certain promoters and activates the QS gene expression in the presence of an autoinducer. Recent research validated this hypothesis by demonstrating that P. aeruginosa isolates expressing mutant LasR genes are not biofilm-producing. 41 Therefore, LasR has gained significant interest as an anti-virulence target.
Over the years, different research groups have identified LasR competitive inhibitors from natural sources, combinatorial chemistry, and computer-based drug designing.19,24,42 Interestingly, P. aeruginosa secretes high concentrations of the phenazine antibiotic pyocyanin, a deep blue pigment, which is believed to be the primary eradicating agent of S. aureus and other bacteria.43,44 Currently, research on the antibacterial property of pyocyanin is exploring its activity against S. aureus and other bacteria, but little or no research is focused on its antibiofilm property. This provides an opportunity to design inhibitors for LasR that can help prevent biofilm production through the hijacking machinery involved and also aid in a decrease in resistance toward antibiotics, pathogenicity, and virulence caused by the production of biofilm as a defense mechanism. The main objective of this study was to find a potent LasR inhibitor through the high-throughput virtual screening approach using HP compounds from the study conducted by 37 Vetrivel et al. explored the antibiofilm activity of some HPs against S. aureus. Compounds for this study were labeled as C1–C9 and HP-14, and the primary scaffold was labeled as HP. Results from the high-throughput virtual screening show that compounds C1–C8 and HP-14 had an allosteric mode of action (see Figure 1). The rest of the compounds did not show any definite binding site, as their binding poses were mainly random across the protein’s surface. The docking score for the allosteric binding compounds ranges from −6.9 to −8.8 kcal/mol (see Table 1). Next, to determine how these binding affinities will affect their mode of action and the stability of their binding interactions, MD simulation studies were carried out with the autoinducer binding at the ligand binding pocket, and this complex was termed the “ternary” complex. Alongside the “ternary” complex is a “binary” complex consisting of AHL and the LasR protein. These complexes were used for further studies and post-MD analysis. A simulation was carried out for 200 ns, which revealed that the compounds formed a stable complex with LasR mediated by favorable interactions with key amino acid residues at the distal site.
Analysis from the ligand RMSD trajectory shows that five HP compounds, C2, C3, C4, C6, and HP-14, were bound at the distal site throughout the simulations (see Figure 3A). Compound C2 established strong interactions with Ser33, Glu133, Lys34, Val232, Ile229, and Arg225. Water-mediated hydrogen bond interactions with the hydroxyl and tertiary nitrogen atoms on the phenazine ring water are also responsible for the stability of the compound. For compound C3, strong interactions were noted with amino acids such as Lys34, Asn55, Ser20, Asn233, Ser28, and Val232. Similar water-mediated hydrogen interactions were also observed. Strong interactions were established between Ser20, Leu177, Leu234, Leu236, Val175, Pro174, Pro57, Arg61, Val53, and Asn55 with compound C4. Amino acid residues responsible for keeping compound C6 at the distal site are Leu234, Leu236, Leu177, Phe219, Val175, Val176, Lys218, Pro170, Pro174, His169, and Lys16. Interestingly, these amino acid residues are found at both the ligand- and DNA-binding domains. This means the compounds acted as pivots between this domain with the aid of these interacting residues, hence leading to structural conformational changes at both domains. In a study by Chowdhury and Bagchi, 45 amino acids such as Glu145, Asp156, Ly42, Thr80, Val232, Arg122, Lys42, Gln25, Met1, Phe210, Thr193, Trp195, Ser194, Lys192, and Ile237 were involved in dimerization through a protein-protein docking approach. 45 In this study, it was revealed that these compounds had interactions with the residues responsible for the dimerization of the LasR protein after the autoinducer is bound to it. This postulates that binding at the distal site will halt the dimerization of the LasR protein. Again Chowdhury and Bagchi 45 identified the solvent-accessible surface area of Gln45, Arg122, and Asp156 in the monomeric and dimeric forms. It was shown that in the dimeric form, these residues elute water to interact with the DNA and are exposed in the monomeric form to aid in dimerization. 45 This phenomenon is also evident in the presence and absence of the autoinducer. In the RMSF analysis, it was noted that the ligand binding caused higher fluctuations in the domains (see Figure 3B). Taking into consideration the effect of compound C3 on the fluctuations of the residues, it was observed that higher fluctuations occurred and caused higher motions of atoms of the amino acid residues, as noted in the PCA analysis of the whole protein structure (see Figure 4). A motion analysis was done in the DNA-binding domain. The PCA of the DNA-binding domain also confirms the motions of the residues at the domain, suggesting that the ligands caused a very intense structural change at the domain (see Figure 5). Supporting the claims by Chowdhury and Bagchi, 45 the results from the PCA study of the DNA-binding domain clearly show that the compounds caused significant motion and structural integrity of the domain because the DNA-binding domain orients in a particular feature to aid in DNA binding (see Figure 5). With this understanding, a critical look into the compactness of the protein is critical. From the Rg study, it was noted that there was an increase in the compactness of the protein on the binding of the compounds at the distal site (see Figure 3C) as compared to the apo and binary complex. Prominent global free energy minima were noted in all complexes, indicating that the structures were stale overall (see Figure 7).

Gibbs free energy landscape. (A) C2-LasR, (B) C3-LasR, (C) C4-LasR, (D) C6-LasR, and (E) HP-14-LasR with the AHL in orange color.
In an attempt to find inhibitors of LasR, researchers have reported several compounds from natural and synthetic routes over the years. These works highlighted and delved more into competitive inhibition, and the compounds reported were shown to be good inhibitors through computational and experimental approaches. The compounds establish strong interactions with residues at the ligand-binding site.8,46,47 Müh et al. proposed that mimicking the interactions of AHL for the design of antagonists would result in the generation of LasR agonists. With this, Geng and co-workers investigated the anti-QS action of luteolin and observed that the compound bound at the active with a dissimilar pattern to that of the AHL. 48 Elsewhere, it has been documented that gliptins have dissimilar binding patterns,19,48 hence luteolin and gliptins are proposed to be effective inhibitors of LasR. Consistent with the literature mentioned above, limited attention has been given to the exploration of non-competitive inhibitors. In this research, the focus was on investigating the allosteric modulation of the LasR protein induced by halogenated compounds, as some of the previously mentioned compounds were found to act as agonists in vitro and in vivo studies. Molecular docking and MD simulations suggested that halogenated compounds have the potential to act as allosteric and non-competitive inhibitors for biofilm inhibition in P. aeruginosa. In addition, binding free energy calculations indicated that the identified compounds bind to the allosteric site through electrostatic and van der Waals interactions, as summarized in Table 2. Based on the study by Huigens et al, 23 HP compounds produce reactive oxygen species (ROS) more than pyocyanin from the experimental evaluation. This suggests that HP compounds show improved eradication of biofilm in gram-positive bacteria and hence would show a similar pattern against gram-negative bacteria strains. Also, the study by Huigens and co-workers highlights that HP compounds are not susceptible to efflux pumps. To support this claim, Huigens and co-workers used a transcriptome analysis of methicillin-resistant S. aureus (MRSA) biofilms treated with HP-14. Results from this analysis show that HP-14 (CLogP = 6.25) rapidly diffuses through bacterial cells and directly binds iron(II) following its release from a siderophore or heme. The binding of iron(II) by HP-14 results in the rapid starvation of iron in MRSA biofilm cells, leading to the activation of iron uptake systems and eventual death, and this finding aligns with a report by Alatawneh and Meijler. 49 Further experimental investigations on these compounds hold a promising strategy for the development of a novel and effective antibiofilm agent against P. aeruginosa.
Conclusions
This study combines molecular docking and MD simulations to understand the molecular mechanisms underlying the anti-biofilm and anti-QS properties of HP compounds. Through virtual screening, all compounds exhibited significant affinity for an allosteric site on LasR, suggesting their potential as lead compounds for further investigation in MD simulations. Among them, five compounds demonstrated stability within the allosteric binding pocket throughout the simulation period, even in the presence of solvent molecules. Interactions between the compounds and amino acid residues at the allosteric site of LasR led to the accumulation of solvent molecules within the binding pocket, potentially leading to protein aggregation. In addition, the compounds perturbed the DNA-binding domain of the protein and interacted with amino acid residues involved in dimerization, thereby inhibiting biofilm formation, as dimerization is crucial for this process. These findings highlight the promising potential of these compounds for further exploration in combating antibiotic resistance across various infections, thus enhancing our ability to address these pervasive health challenges. Future research can focus on in vitro and in vivo experimental evaluation of these compounds against the LasR. Other researchers can help expand scientific data through Quantitative Structure Activity Relationship (QSAR) studies.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
PBN, PM, and AK conceived the study. All experiments were designed by PBN, PM, POP and AK. Computations were made by PBN, PM, POP and AK. Data analysis was done by PBN, PM, POP and AK. The initial manuscript draft was prepared by PBN, PM, POP and AK. All authors read and approved the final manuscript.
Data Availability
All data generated or analyzed during this study are included in this published article.