
Abstract
Nucleotide base composition plays an influential role in the molecular mechanisms involved in gene function, phenotype, and amino acid composition. GC content (proportion of guanine and cytosine in DNA sequences) shows a high level of variation within and among species. Many studies measure GC content in a small number of genes, which may not be representative of genome-wide GC variation. One challenge when assembling extensive genomic data sets for these studies is the significant amount of resources (monetary and computational) associated with data processing, and many bioinformatic tools have not been optimized for resource efficiency. Using a high-performance computing (HPC) cluster, we manipulated resources provided to the targeted gene assembly program, automated target restricted assembly method (aTRAM), to determine an optimum way to run the program to maximize resource use. Using our optimum assembly approach, we assembled and measured GC content of all of the protein-coding genes of a diverse group of parasitic feather lice. Of the 499 426 genes assembled across 57 species, feather lice were GC-poor (mean GC = 42.96%) with a significant amount of variation within and between species (GC range = 19.57%-73.33%). We found a significant correlation between GC content and standard deviation per taxon for overall GC and GC3, which could indicate selection for G and C nucleotides in some species. Phylogenetic signal of GC content was detected in both GC and GC3. This research provides a large-scale investigation of GC content in parasitic lice laying the foundation for understanding the basis of variation in base composition across species.
Keywords
Introduction
Genomic characteristics, such as base composition, play an important role in the evolution and ecology of organisms. These features can be influential in molecular mechanisms involved in gene function, phenotype, and amino acid composition.1-3 Base composition is typically measured as GC content (proportion of guanine and cytosine in DNA), which has been directly linked to amino acid composition. 4 As amino acids are the building blocks of proteins, variation in amino acid composition is a critical component of protein evolution. 4 Measuring GC content within and among species may be the first step for understanding adaptation at a molecular level. For example, identifying adaptive alleles, which may be indirectly constrained based on GC content,5,6 in threatened populations is imperative for species and ecosystem conservation. 7 GC content has been found to be highly variable among and within species as well as at different organizational levels (ie, proteome vs genome8,9). Here, we mainly focus on the GC content of protein-coding genes, and it is important that comparisons between studies be made using analogous data sets. Even with this large-scale variability, some patterns stand out, such as higher recombination rates in GC-rich genes 10 and a negative relationship between GC content of third codon positions and chromosome length.11,12 The patterns of GC content variation across organisms are thought to be linked to genomic characteristics such as methylation throughout the genome, 13 expression levels of coding genes14,15, and genome-wide gene conversion. 16 Many hypotheses have been suggested to explain variation in GC content across different regions of the genome, such as molecular mechanics, environmental factors, natural selection, or a combination thereof.16-18 Before any of these mechanisms can be investigated, determining GC content across an organism’s genes and comparing the variation found among closely related species is needed to understand how base composition has influenced diversification and adaptation. 19
A disproportionate amount of genomic research has focused on vertebrate groups, particularly mammals and birds.20,21 Insects have been given much less attention yet are the most diverse group of animals in the world 22 and are facing catastrophic declines. 23 Parasitic lice (Phthiraptera) are of particular interest due to their global distribution, fast rate of evolution, and high level of diversification,24-28 providing an ideal system to study GC content and protein evolution in a closely related group of organisms 29 . Lice have among the smallest genomes within insects 30 ; however, only a few studies have examined GC content of coding genes in this group with conflicting results. A small number of genes were found to be GC-rich, whereas a much larger set were GC-poor.31,32 Many insects have low GC content overall,33-36 which is consistent with the results of GC content of coding genes in parasitic lice found by Virrueta Herrera et al, 32 even though the mtDNA of some parasitic lice genes is GC-rich compared with other insects31,37. A large-scale genome-wide investigation would provide a better understanding of the patterns in base composition of parasitic lice and allow for more comprehensive comparisons with other organisms.
These genomic studies often use gene assembly programs run on a high-performance computing (HPC) cluster. A significant amount of computational resources are necessary for large-scale data, and these resources are not always available or accessible. 38 Researchers often pay for an allotted amount of resources and are charged for these resources even if they are idle, leading to wasted time and money. 39 Given that gene assemblies are generally resource-intensive, inefficient resource use can quickly become wasteful. A critical component of continuing the advancement and accessibility of molecular studies is the development of programs that can efficiently prepare and analyze these genome-scale data sets.
Automated target restricted assembly method (aTRAM)40,41 is a targeted gene assembly program, which assembles specific loci from unassembled sequences using a closely or distantly related locus as the reference. 42 Automated target restricted assembly method begins by creating a library from the unassembled sequence reads, which consists of BLAST formatted databases split into multiple groups of paired-end sequences and a relational database to associate read-pairs. Next, a target sequence is blasted against these groups to identify homologous reads which are then assembled into a contiguous piece of DNA, termed contig. This process is repeated using the newly assembled contig as the query sequence and so on for multiple iterations until the target locus is assembled. Through this process, aTRAM breaks up large tasks (eg, Basic Local Alignment Search Tool (BLAST)) into multiple small tasks, using central processing units (CPUs). Automated target restricted assembly method allows the user to determine the number of CPUs in each run and uses them to blast the groups of paired-end reads within a library in parallel, increasing the overall rate of gene assembly. However, it is unclear if increasing the number of CPUs results in the most efficient use of resources. Adding more CPUs might be lowering the computational efficiency as more of these resources might be sitting idle.
Here, we assembled all nuclear protein-coding genes for several feather louse (Ischnocera) genera. We used a reference set of genes from the recently annotated pigeon louse (Columbicola columbae) genome 43 to measure GC content across the proteome of this diverse group of ectoparasites. We compared GC content among genes and across species and estimated the phylogenetic signal of GC content. Our aim was to gain an understanding of the level of variation in base composition found within this group of insects and to provide the data needed to continue investigating protein evolution.
We used aTRAM, which was designed to maximize resource use by allowing the user to incorporate as many cores as available during the assembly process. However, based on the time and resources used to assemble a large number of genes from a single taxon, it is unclear if aTRAM is using those resources efficiently, or if there are more optimal ways to use resources (eg, in parallel) to maximize efficiency. Before assembling the genes for our full set of taxa, we first investigated aTRAM resource efficiency given different amounts of computational resources to see how the available resources are being used during the assembly process. Focusing on 2 of the most commonly manipulated computational resources (CPUs and tasks), we measured the rate of and computational efficiency of gene assemblies using a varying number of resources. Our goal is to maximize computational efficiency whereas optimizing the rate at which genes can be assembled.
Methods
Our data set included 57 species in 54 genera of parasitic feather lice. Raw data were obtained from the NCBI (National Center for Biotechnology Information) Sequence Read Archive (SRA) (see Supplemental Data for SRA). All paired-end reads were trimmed using Trimmomatic v0.39 in paired-end mode to remove areas of low quality and to clip adapters (Illumina universal adapter). 44 We used a sliding window of 4 base pairs with a minimum quality of 20 (Phred + 33), and all reads shorter than 100 base pairs were dropped. All genes were assembled with aTRAM v2.4.3 42 using the 13 362 annotated genes from the pigeon louse genome 43 as a reference.
aTRAM
The software aTRAM was designed to maximize computational resources. For example, running BLAST searches on each group of paired-end reads can be done in parallel using many CPUs or sequentially using a single CPU. It is unknown which of the following methods would improve gene assembly rate and resource efficiency: a) adding more CPUs linearly or b) running multiple instances of aTRAM in parallel with fewer CPUs. Although HPC clusters generally have multiple options for resource manipulation, we focus on 2 common resources users have to select when running programs with HPC: numbers of CPUs and tasks. All computational tests were run on the University of Nevada, Reno HPC cluster, Pronghorn, which uses Slurm as a workload manager. For all tests, the same gene (chr1-aug-0.14-mRNA-1) and library (Alcedoecus: 55 groups of paired-end reads) were used. We examined resource efficiency by measuring the percent of available resources used with an increasing scale of CPUs and tasks.
CPUs
We altered Slurm
Tasks
The next test focused on parallelization to measure CPU use efficiency and gene assembly rate. All tests were run exclusively on a single node with Slurm argument
Full data set assembly
We assembled a target set of 13 364 of the protein-coding genes for 57 feather lice taxa based on the results from the CPU and task tests with aTRAM using ABySS. These genes were all of the annotated genes from the pigeon louse genome. The amino acid sequences from these genes were used as the reference for tblastn searches. Exonerate 47 was used to stitch together assembled contigs and concatenate exons using the pigeon louse amino acid reference sequences. Once genes were assembled, genes that were not selected as reciprocal best hit (RBH) for the associated pigeon louse target after a reciprocal best BLAST search were removed. Using the pigeon louse gene set, we ran analysis of variance (ANOVA) tests in R v4.3.1 48 to compare the length of all genes, the length of genes that assembled for at least 1 taxon, and the length of genes that did not assemble for any taxa.
GC content
GC content was measured
Phylogenetic signal
We tested for phylogenetic signal of GC and GC3 to see whether closely related feather lice taxa have similar GC content compared with more distant relatives. Analyses were done using the phylosignal v1.3 55 and phylobase v0.8.10 56 packages in R. We first measured global phylogenetic signal across the entire phylogeny with both Moran I and Abouheif Cmean with 100 simulations and 999 repetitions. Moran I and Abouheif Cmean are measures of spatial autocorrelation used to test the level of similarity of a characteristic between branch tips that are close in proximity.57,58 Abouheif Cmean is a slight variation of Moran I by ignoring branch lengths and focusing on the mean of multiple topology possibilities with a weighted matrix of relatedness. 59 This accounts for any inaccuracies in the tree and when paired with Moran I can provide more confidence that any signal found is not reliant on the estimated branch lengths. In addition, Abouheif Cmean is a robust analysis that provides accurate statistics under many conditions (ie, polytomies or tree size 60 ). Both indices calculate a value from −1 to 1, meaning the absence (−1) or presence (1) of similarities in a trait at certain phylogenetic distances. Second, because global measures of phylogenetic signal do not specify which lineages show trait similarities, we ran a Local Indicators of Phylogenetic Association (LIPA) analysis to identify local hotspots of autocorrelation. 55
Results
Central processing units
Trinity and ABySS had similar patterns of gene assembly rate and resource efficiency, as measured by genes assembled per CPU per hour, although ABySS assembled genes faster than Trinity overall (Figure 1A and B). We found that aTRAM used computational resources most efficiently (ie, assembled the most genes per CPU) when given 1 CPU (Trinity: 7 genes/CPU/h—ABySS: 8 genes/CPU/h; Table 1). This is a common outcome for most parallelization problems, as any increase in parallelization introduces overhead in the form of thread communication and synchronization. When addressing parallelization of aTRAM using tasks, we used the Slurm argument

Gene assembly rate (A and C) and resource use efficiency (B and D) across an increasing number of CPUs (A and B) and aTRAM instances (C and D). An instance in this context represents a task, specifically the number of concurrently running aTRAM instances. Two different assemblers were used: ABySS (pink circles) and Trinity (blue triangles).
Statistics from assembling genes with an increasing number of CPUs on a high-performance computing cluster using 2 different assemblers (Trinity and ABySS).

Each row indicates a single run and how many CPUs were used. Resource efficiency was calculated by the number of genes assembled per available CPU per hour.
Tasks
For both assemblers, 32 tasks resulted in the highest gene assembly rate and CPU efficiency (Trinity: 1.60 genes/min and 22.04% CPU efficiency; ABySS: 2.29 genes/min and 35.23% CPU efficiency; Table 2). Speedup
Statistics from assembling genes with an increasing number of tasks on a high-performance computing cluster using 2 different assemblers (Trinity and ABySS).

Each row indicates a single run and how many tasks were used. All runs were given 1 CPU per task based on the results from scaling CPUs. Speedup measures the relative improvement of the same run with differing resources. Central processing unit and memory efficiency were obtained from the workload manager (Slurm) output (CPU efficiency = cpu_time / (run_time × number_of_cpus); memory efficiency is the amount of allocated memory used in a run).
Full data set assembly
Based on the results from the CPU and task scaling tests, we assembled all of the protein-coding genes from 57 feather lice taxa using the following Slurm arguments:
Within our data set, 11 780 genes assembled for at least 1 taxon and 7182 of those genes were assembled by a minimum of 54 taxa. We found that 1584 genes did not assemble for any taxon. Using the nucleotide sequences from the pigeon louse reference, we compared gene length between all of the reference genes (n = 13 364), genes that we assembled for at least 1 taxon (n = 11 780), and genes that did not assemble for any taxon (n = 1584). Data were log transformed to fit with assumptions of normality. Gene lengths between these groups were significantly different with a large effect size showing shorter genes were less likely to assemble (ANOVA: F2, 26 726 = 2270, P = < .001, η2 = 0.15). Pairwise t tests with Bonferroni correction showed a significant difference between all group pairings (P ⩽ .001; Figure 2).

Differences in gene length (measured in DNA) from Columbicola columbae between 3 groups: all C. columbae protein-coding genes (n = 13 364; green), genes that assembled for at least 1 taxon in our data set (n = 11 780; pink), and genes that did not assemble for any taxon (n = 1584; yellow). The group of genes that did not assemble exhibited significantly shorter gene lengths compared with the other 2 groups. The presented data use logged values, with the y-axis indicating non-logged values.
GC content
We measured GC content for all genes for each taxon (n = 499 426), as well as GC content for each codon position (GC1, GC2, and GC3). Data followed a normal distribution for GC, GC1, and GC2. The distribution for GC3 was generally normal with a long right tail, indicating a disproportionate group of genes that are GC-rich. The range of GC content for all genes was 19.57% to 73.33% (M = 42.96%; SD = 6.53; see Supplemental Data for all measures of GC content per taxon). Only 15% of all genes in our data set had a GC content of 50% or greater with most genes being GC-poor. Overall GC content was significantly different between taxa (n = 499 426; ANOVA: F56, 496 925 = 1410, P ⩽ .001) with a large effect size (η2 = 0.14). To be sure the significance of the ANOVA was not due to only a few groups with significant differences, we ran a pairwise t test with Bonferroni correction and found a significant difference in GC content between 91% of groups (P ⩽ .05). The mean GC content across all protein-coding genes for the pigeon louse reference (n = 13 364) was 41.40% (SD = 5.17), similar to the GC content found in our data set. Both of these findings, however, are higher than GC content found across the entire pigeon louse genome, which is 36%. 42 For each of the 3 codon positions, mean GC content was 47.80% (SD = 5.32) for GC1, 37.70% (SD = 5.96) for GC2, and 43.37% (SD = 14.15) for GC3. As expected, there was much more variation found at GC3 compared with GC1 and GC2. The distribution of GC content per taxon for GC, GC1, GC2, and GC3 is shown in Supplemental Figures 1 to 4.
We found a positive correlation between GC content and variation in GC content (measured as SD) for overall GC (r = 0.61, P ⩽ .001; Figure 3) and for GC3 (r = 0.70, P ⩽ .001; Figure 3). Species with higher GC content have significantly more variation across their genes, whereas those that are more GC-poor seem to have a more consistent base composition. We found a weak positive relationship between GC content and gene length (ANOVA: F(1, 496 980) = 4355, P ⩽ .001; η2 = 0.0087) among the genes. There is a slightly stronger positive relationship between GC and gene length for the pigeon louse reference; however, the effect size is still quite small (ANOVA: F(1, 13 362) = 158.2, P ⩽ .001; η2 = 0.01).
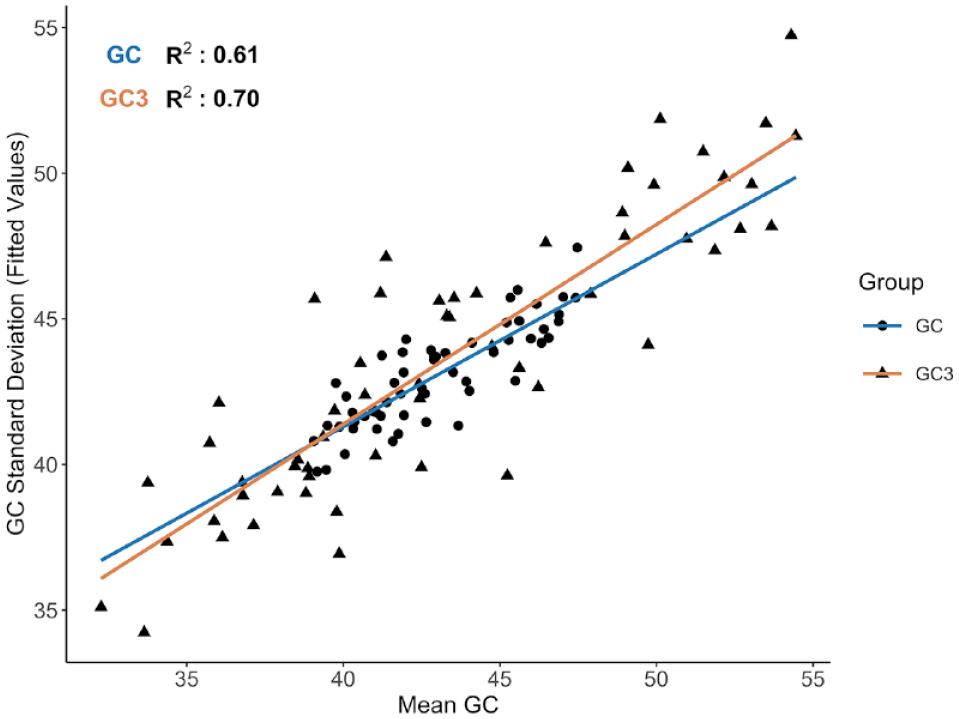
A strong positive correlation was found between both mean GC (blue, circles) and GC3 (GC content at codon position 3; orange, triangles) and their respective SDs. Although accounting for phylogeny, R2 values were 0.61 for GC and 0.70 for GC3. Mean GC, GC3, and SD were calculated for all genes assembled per taxon. Each data point represents a distinct genus, and regression lines are depicted in blue (GC) and orange (GC3). The y-axis shows the fitted values calculated from the phylogenetic linear regression model.
GC content on tree and phylogenetic signal
GC content was mapped onto the subsampled phylogenetic tree from de Moya et al, 28 and in some cases, related taxa had similar GC levels (Figure 4). The pattern of GC content seen across the tree is also seen at all codon positions (Supplemental Figures 5 and 6), specifically for GC3 (Supplemental Figure 7). This pattern suggests that transitions between GC-rich and GC-poor genes may have occurred multiple times across this group, as opposed to a more continuous change from older to more recently diverged taxa (see 19 ).

Phylogenetic tree (modified from de Moya et al 28 ) with mean GC content mapped onto branches. Local hotspots exhibiting significant phylogenetic signal, as determined by the LIPA analysis, are indicated by asterisks. Purple circles represent a positive relationship, whereas orange rectangles denote a negative relationship. Darker shades of blue indicate higher GC levels, whereas lighter shades of green represent lower GC levels.
Phylogenetic signal for GC and GC3, was not found using Moran I but was when using Abouheif Cmean. For overall GC, Moran I was insignificant and close to 0 (I = −0.0099, P = .100) whereas Abouheif Cmean was significant with a low positive signal (CM = 0.190, P = .020). A similar pattern appeared for GC3 with no global phylogenetic signal detected with Moran I (I = −0.010, P = .077) but this signal was detected with Abouheif Cmean (CM = 0.194, P = .015). Although no significant phylogenetic signal was found regarding GC using Moran I at the global scale, the LIPA analysis found significant positive autocorrelation for 13 taxa using Moran I, as well as for 15 taxa using Abouheif Cmean (Figure 4). Two of the taxa identified as having significant phylogenetic signal with Abouheif Cmean were negatively autocorrelated, whereas all of the taxa that exhibited significant phylogenetic signal using Moran I were positively autocorrelated. For GC3, the LIPA analysis for both Moran I and Abouheif Cmean revealed significant autocorrelation for the same 14 taxa; however, 2 of these were negatively autocorrelated with Abouheif Cmean (Supplemental Figure 7).
Discussion
Considering the disproportionately lower amount of genetic research on insects relative to their abundance and global distribution, 20 we set out to accomplish 2 main goals. First, we aimed to improve gene assembly efficiency with aTRAM by decreasing the amount of time and resources needed using parallelization with HPC, allowing for easier access to genetic data for these understudied groups. Second, we investigated the base composition of the largest set to date of protein-coding genes of feather lice. We found that while 32 CPUs assembled the most genes per minute, resource use was most efficient when using 1 CPU (Trinity: 7 genes/CPU/ hr| ABySS: 8 genes/CPU/h; Figure 1A and B). Using a single CPU, the number of tasks that offered the highest rate of genes assembled per minute, as well as the best CPU and memory efficiency was 32 tasks (Trinity: 1.6 genes/min| ABySS 2.29 genes/min; Figure 1C and D). By manipulating the resources given to aTRAM, we obtained a gene assembly speedup of 9.10 (Trinity) and 12 (ABySS; Table 2) times. This reduced the assembly time of a set of 13 364 genes from 48.72 to 3.8 days. As seen in our analysis, increasing the number of CPUs does not necessarily improve efficiency of a program (Figure 1B). This becomes increasingly complicated when addressing additional components of HPC, such as RAM, permanent disk space, and threading. In general, we recommend running similar tests on other software to gain a general understanding of a program’s resource usage with an HPC system.
Our final data set included 499 426 genes and on average had a GC content below 50% (mean GC = 42.96%; SD = 6.53). For third codon positions only, we found a similar average GC3 to that found by Virrueta Herrera et al; 32 however, we found a higher average GC1 and lower average GC2. We also found higher variation in GC content (19.57%-73.33%) across all genes. One explanation for the increased GC content in our data set compared with Baldwin-Brown et al 43 is the bias toward easily aligned genes. We retained only those genes that could be identified by reciprocal best-hit BLAST to the pigeon louse genome. These genes are less likely to contain repetitive sequences, and repetitive genome features are known to be GC-poor. By only retaining easily aligned genes, we have likely removed GC-poor genes from the data set.
Our results suggest feather lice may have higher GC content in coding sequences compared with many other insects that have been investigated. For example, GC content of protein-coding genes from 2 species of parasitoid wasp is around 30% (Aphidius ervi and Lysiphlebus fabarum 36 ) and between 33% and 39% for the honeybee (Apis mellifera 62 ). Some species of insects do have similar GC content to feather lice, such as silkworms (Bombyx 63 ). Unfortunately, the genetic studies that focus on insects are overrepresented by a small number of model species, such as members of Drosophila and Lepidoptera,20,64 ignoring the enormous diversity of insects. To properly understand how GC content of feather lice compares to other insects, more studies are needed on non-model species.
For overall GC and GC3, we found a significant correlation between %GC for a species and SD among genes within that species (Figure 3), a pattern also seen in many vertebrates. 11 Species with higher values for GC and GC3 showed much more variation compared with species with lower GC and GC3. This could indicate that GC-rich genes are being selected for in some species, resulting in more GC variation across their genes. Alternatively, this could be a consequence of GC-biased gene conversion (gBGC), which favors the use of G and C bases. 65 This is particularly prominent in species with high rates of recombination. When an allelic mismatch occurs during the repair of a meiotic double-strand break, gBGC results in a biased frequency of G:C compared with A:T conversions, thus increasing the chance of GC substitution.10,66 It is thought that gBGC plays a significant role in the variation in GC content within genomes of some taxonomic groups. 67 Many studies have found evidence that suggests gBGC plays a prominent role in the GC-rich base composition of avian 68 and mammalian isochores.69,70 In addition, Pessia et al 71 examined the genomes of a broader range of organisms (Unikonts, Excavates, Chromalveolates, and Plantae) and found that gBGC is evident in most eukaryotic groups. It is important to note that disentangling gBGC from directional selection requires further analysis. Although the mechanism is different, both result in a biased increase in 1 allele. 72 Because feather lice have low GC content and high rates of substitution, it may be that gBGC is not a strong influence on GC content in this group and other factors play a larger role. Alternatively, these results could indicate a shift toward higher GC heterogeneity, where different mechanisms result in GC-rich and GC-poor regions of the genome (eg, 73 ).
Another prominent hypothesis that could also explain GC variation and evolution focuses on selection acting on the molecular machinery that results in nucleotide biases. Specifically, deamination of methyl-cytosine produces thymine causing a T:G mismatch requiring repair. 74 Methyl-cytosine deamination and GC content create a positive feedback loop that can either decrease or increase GC content in an organism. 75 In addition, some studies have found a positive correlation between GC content of transposable element (TE) and genome-wide GC content (eg, 76 ). GC-rich TEs can increase GC content within the region of insertion if that region has a lower %GC than the TE, and the same rule would follow for GC-poor TEs. However, parasitic lice have a relatively low percentage of TEs in their genome (<5%)42,77,78. Interestingly, this fraction of genome-wide TEs is lower than that of silkworms (~40% of genome made up of TEs 79 ), with whom feather lice share a similar average GC content. 73 Continuing to investigate the mechanisms driving GC content and the evolutionary consequences of GC-rich or poor genomes is critical for deepening our understanding of molecular evolution and, ultimately, species diversification.80,81 To determine the driving force behind changes in base composition of feather lice among other organisms, future research needs to focus on a combination of synonymous vs non-synonymous substitutions (dN/dS ratios), 82 effective population size (Ne), 83 and codon usage bias. 84
Phylogenetic signal indicates groups of related organisms that may exhibit similar ecological or genetic traits 85 because of phylogenetic relatedness alone. We estimated the phylogenetic signal of GC content in feather lice, which revealed that close relatives had more similar GC content than would be expected by chance. Specifically, local phylogenetic signal of GC content was detected for overall GC (Figure 4) and GC3 (Supplemental Figure 7). In addition, using Abouheif Cmean, we identified positive global phylogenetic signal of mean GC and GC3 (GC: CM = 0.190, P = .020; GC3: CM = 0.194, P = .015). The LIPA analysis found 15 (GC) and 14 (GC3) species with significant autocorrelation, most being positive. Thus, GC content is likely a feature of phylogenetically closely related lineages. Because of this, we can have more confidence that base composition alone will not bias the construction of phylogenetic trees, a common concern in systematics. 86
Feather lice genera have been grouped into different ecomorphs based on similar morphological characteristics. These specialized phenotypes allow them to live in different areas of their avian host’s body to escape host defense mechanisms 24 ,87-89 and found evidence of convergent evolution in these ecomorphs but it is unknown if each ecomorph type experiences the same selective pressures on the same genes or expresses similar genetic pathways. Signatures of convergent evolution have been found in other species that exhibit ecomorphs, which can tell us more about the genetic architecture of closely and distantly related organisms and the role selective pressures play in diversification.90,91 Furthermore, these data provide new opportunities for future studies to explore insect protein evolution and adaptive evolution between feather lice genera and species. 92
Scientists without a background in bioinformatics or computer science, however, often struggle with using the programs necessary for these evolutionary studies. Because this software often needs to be executed with HPC, researchers frequently hire specialists if they have available funds. This burden is much heavier on minorities, as they are less likely to receive funding93,94. These programs need to be built in a way that is more accessible and resource-efficient when used by scientists at large. Future development should increase focus on improving the program’s ability to identify and allot available computational resources on various HPC architectures without the need for complex manipulation by the user. We have increased accessibility to aTRAM by developing a Singularity container that provides a more simple manner to install and use the program (https://github.com/averygrant/atram_singularity). Our aTRAM container consists of all of the files and dependencies needed for running aTRAM leading to fewer installation steps.
Genomic base composition is a fundamental feature of the genome of all organisms. Our results show that most feather louse protein-coding genes are GC-poor with the greatest variation found in GC3. However, GC content varies considerably between species. On average, lice have a higher GC content than other insects, although there is considerable variation in GC content among insect species. For example, feather lice GC content is similar to silkworms, 63 while being very different from the pea aphid (Acyrthosiphon pisum). 33 More detailed comparisons of GC content among insects will require examination of this feature at a genomic scale, ideally comparing orthologous genes. It is expected that GC content may vary between different gene regions,31,95 such as nuclear vs mtDNA96,97 and introns vs exons, 98 and this variation needs to be taken into account. As insect genomes become more available for a higher diversity of species, more research can focus on untangling the impact of molecular mechanisms and selective pressures on GC-rich or GC-poor organisms and, ultimately, shed light on protein evolution in insects.
Supplemental Material
sj-png-2-bbi-10.1177_11779322241257991 – Supplemental material for Rapid Targeted Assembly of the Proteome Reveals Evolutionary Variation of GC Content in Avian Lice
Supplemental material, sj-png-2-bbi-10.1177_11779322241257991 for Rapid Targeted Assembly of the Proteome Reveals Evolutionary Variation of GC Content in Avian Lice by Avery R Grant, Kevin P Johnson, Edward L Stanley, James Baldwin-Brown, Stanislav Kolenčík and Julie M Allen in Bioinformatics and Biology Insights
Supplemental Material
sj-png-3-bbi-10.1177_11779322241257991 – Supplemental material for Rapid Targeted Assembly of the Proteome Reveals Evolutionary Variation of GC Content in Avian Lice
Supplemental material, sj-png-3-bbi-10.1177_11779322241257991 for Rapid Targeted Assembly of the Proteome Reveals Evolutionary Variation of GC Content in Avian Lice by Avery R Grant, Kevin P Johnson, Edward L Stanley, James Baldwin-Brown, Stanislav Kolenčík and Julie M Allen in Bioinformatics and Biology Insights
Supplemental Material
sj-png-4-bbi-10.1177_11779322241257991 – Supplemental material for Rapid Targeted Assembly of the Proteome Reveals Evolutionary Variation of GC Content in Avian Lice
Supplemental material, sj-png-4-bbi-10.1177_11779322241257991 for Rapid Targeted Assembly of the Proteome Reveals Evolutionary Variation of GC Content in Avian Lice by Avery R Grant, Kevin P Johnson, Edward L Stanley, James Baldwin-Brown, Stanislav Kolenčík and Julie M Allen in Bioinformatics and Biology Insights
Supplemental Material
sj-png-5-bbi-10.1177_11779322241257991 – Supplemental material for Rapid Targeted Assembly of the Proteome Reveals Evolutionary Variation of GC Content in Avian Lice
Supplemental material, sj-png-5-bbi-10.1177_11779322241257991 for Rapid Targeted Assembly of the Proteome Reveals Evolutionary Variation of GC Content in Avian Lice by Avery R Grant, Kevin P Johnson, Edward L Stanley, James Baldwin-Brown, Stanislav Kolenčík and Julie M Allen in Bioinformatics and Biology Insights
Supplemental Material
sj-png-6-bbi-10.1177_11779322241257991 – Supplemental material for Rapid Targeted Assembly of the Proteome Reveals Evolutionary Variation of GC Content in Avian Lice
Supplemental material, sj-png-6-bbi-10.1177_11779322241257991 for Rapid Targeted Assembly of the Proteome Reveals Evolutionary Variation of GC Content in Avian Lice by Avery R Grant, Kevin P Johnson, Edward L Stanley, James Baldwin-Brown, Stanislav Kolenčík and Julie M Allen in Bioinformatics and Biology Insights
Supplemental Material
sj-png-7-bbi-10.1177_11779322241257991 – Supplemental material for Rapid Targeted Assembly of the Proteome Reveals Evolutionary Variation of GC Content in Avian Lice
Supplemental material, sj-png-7-bbi-10.1177_11779322241257991 for Rapid Targeted Assembly of the Proteome Reveals Evolutionary Variation of GC Content in Avian Lice by Avery R Grant, Kevin P Johnson, Edward L Stanley, James Baldwin-Brown, Stanislav Kolenčík and Julie M Allen in Bioinformatics and Biology Insights
Supplemental Material
sj-png-8-bbi-10.1177_11779322241257991 – Supplemental material for Rapid Targeted Assembly of the Proteome Reveals Evolutionary Variation of GC Content in Avian Lice
Supplemental material, sj-png-8-bbi-10.1177_11779322241257991 for Rapid Targeted Assembly of the Proteome Reveals Evolutionary Variation of GC Content in Avian Lice by Avery R Grant, Kevin P Johnson, Edward L Stanley, James Baldwin-Brown, Stanislav Kolenčík and Julie M Allen in Bioinformatics and Biology Insights
Supplemental Material
sj-xlsx-1-bbi-10.1177_11779322241257991 – Supplemental material for Rapid Targeted Assembly of the Proteome Reveals Evolutionary Variation of GC Content in Avian Lice
Supplemental material, sj-xlsx-1-bbi-10.1177_11779322241257991 for Rapid Targeted Assembly of the Proteome Reveals Evolutionary Variation of GC Content in Avian Lice by Avery R Grant, Kevin P Johnson, Edward L Stanley, James Baldwin-Brown, Stanislav Kolenčík and Julie M Allen in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
The authors thank Sebastian Smith and John Anderson for their help with the computational components of this publication.
Author Contributions
ARG and JMA conceived and designed the study; ARG performed the formal analyses, created all visualizations, and wrote the original draft; all authors contributed to data collection, and reviewing and editing; ARG, JMA, and KPJ analyzed results; JMA supervised and funded the research; all authors approved the final manuscript.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by NSF grants 1925312 to JMA, NSF DEB 1925487 and DEB 1926919 to KPJ. The computational work in this publication was made possible by a grant from the National Institute of General Medical Sciences (GM103440) from the National Institutes of Health. The authors would like to acknowledge the support of Research & Innovation and the Cyberinfrastructure Team in the Office of Information Technology at the University of Nevada, Reno for facilitation and access to the Pronghorn High-Performance Computing Cluster.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.