
Abstract
The activation of the Wnt signaling pathway is implicated in a neuroprotective mechanism against the Alzheimer disease. When this pathway is blocked, it activates GSK3 beta, leading to tau hyperphosphorylation and the apoptosis of neurons. Dickkopf-related protein 1 (DKK1) is a protein that competes with the Wnt ligand for the low-density lipoprotein receptor–related protein 6 (LRP6) receptor’s binding, interrupting the Wnt-induced Fzd-Wnt-LRP6 complex. This counteracts Wnt’s neuroprotective effect and contributes to the progression of the Alzheimer disease. The aim of this study was to use in silico approach to develop new agents that can combat the Alzheimer disease by targeting the interaction between DKK1 and LRP6. To achieve this, we conducted a virtual screening (Vsw) of the Asinex-CNS database library (n = 54 513) compounds against a generated grid in LRP6 protein. From this screening, we selected 6 compounds based on their docking score and performed molecular mechanics-generalized Born surface area (MM-GBSA) binding energy calculations on the selected ligands. Next, we evaluated the Absorption, Distribution, Metabolism, and Excretion (ADME) results of the 6 screened compounds using the Quick prop module of Schrödinger. We then employed several computational techniques, including PCA (Principal Component Analysis), DCCM (Dynamic Cross-Correlation Map), molecular dynamics simulation, and molecular mechanics/Poisson-Boltzmann surface area (MM/PBSA)–based negative binding free energy (BFE) calculation, to further analyze the compounds. Our extensive computational analysis resulted in the identification of 3 potential hits, LAS 29757582, LAS 29984441, and LAS 29757942. These compounds were found to block the interaction of DKK1 with LRP6 (A and B interface) protein, and their potential as therapeutic agents was supported by negative BFE calculation. Therefore, these compounds show potential as possible therapeutic agents for treating the Alzheimer disease through targeting the interaction between DKK1 and LRP6.
Introduction
The Alzheimer disease (AD) is common form of dementia, which primarily affects the aging population. Its exact cause is still unknown. The pathophysiological mechanism of AD is characterized by the deposition of the aggregated form of amyloid-β (Aβ) peptide and hyperphosphorylated tau proteins. Numerous signaling pathways have been associated with AD, and the Wnt signaling pathway is increasingly becoming important in drug-target investigations. 1 In the Wnt signaling pathway, a variety of Wnt (wingless integrated) molecules aid in activating the pathway by stimulating extracellular signaling to several intra-cellular signal transduction cascades, including the Wnt/beta-catenin-dependent and independent pathways. 2 In the Wnt/beta-catenin-dependent signaling pathway, the 2 co-receptors are serpentine transmembrane Fz (Frizzled) receptors and the second is a single pass LRP5/6 co-receptor (low-density lipoprotein receptor–related protein) for binding of Wnt ligands. The LRP5/6 co-receptor protein consists of 2 propellers: LRP6E1-E2 and LRP6 E3-E4. The N-terminal region E1-E2 binds to Wnt9B and the E3-E4 region binds to Wnt3A. 3 Wnt ligands activate the cytoplasmic tail region of LRP5/6 through phosphorylation, which subsequently activates the Disheveled (Dsh/Dvl) protein. This scaffold protein is responsible for degrading the destruction complex, including GSK3beta (glycogen synthase kinase 3 beta), CK1 (casein kinase 1), APC (axis inhibition protein), and AXIN. This leads to increased beta-catenin levels, which acts as a transcription factor to activate downstream genes necessary for cell survival. Disturbances in the Wnt signaling pathway can result in a wide range of diseases, including cancer, neurodegenerative diseases, bone disorders, and other conditions. However, the activation of the Wnt/beta-catenin signaling pathway has been shown to have a role in neuroprotection mechanisms. 1 In the case of AD, inhibition of the Wnt signaling pathway is associated with GSK3 beta activation, as reported previous studies. This activation further leads to hyperphosphorylation of the tau protein, 4 resulting in tangle formation and eventually neuronal apoptosis.4-6 However, when β-catenin protein is phosphorylated by the activated destruction complex, it is sent for ubiquitylation and degradation. As a result, β-catenin is unable to reach the nucleus to express the survival genes, leading to neuronal degeneration, which subsequently hampers the process of learning and memory (Figure 1). Therefore, the Wnt/β-catenin signaling pathway plays an important role in AD. 1 There are several agents that can attenuate the Wnt signaling pathway, one of which is Dickkopf (DKK), a competitive inhibitor protein for LRP5/6 receptor. 3 This protein is found in both bone and brain and binds to the extracellular region of LRP, disrupting the Wnt3A/Fzd/LRP6 complex.7,8 Dickkopf protein is a secreted glycoprotein which has 4 members—DKK1, DKK2, DKK3, and DKK4.9-11 Dickkopf-related protein 1 has 2 cysteine-rich regions (CRD1 and CRD2) that interact with the third and fourth propellers (E3 and E4) of LRP6, and this interaction inhibits Wnt signaling. The complex of the C-terminal of DKK1 protein and LRP6 receptor protein has been validated by size exclusion chromatography.7,9,11 There are 2 proposed mechanisms for how DKK1 opposes LRP5/6. The first mechanism suggests that DKK1 disrupts the Wnt-induced Fzd-Wnt-LRP6 complex primarily by binding LRP5/6, leading to inhibition of Wnt/β-catenin signaling. The second mechanism proposes that DKK1 and Kremen trigger LRP5/6 endocytosis and degradation in specific tissues. Modifying the interactions between DKK and LRP5/6 could be a useful therapeutic approach for various diseases, including multiple myeloma, osteoporosis, osteosarcoma, and melanoma.
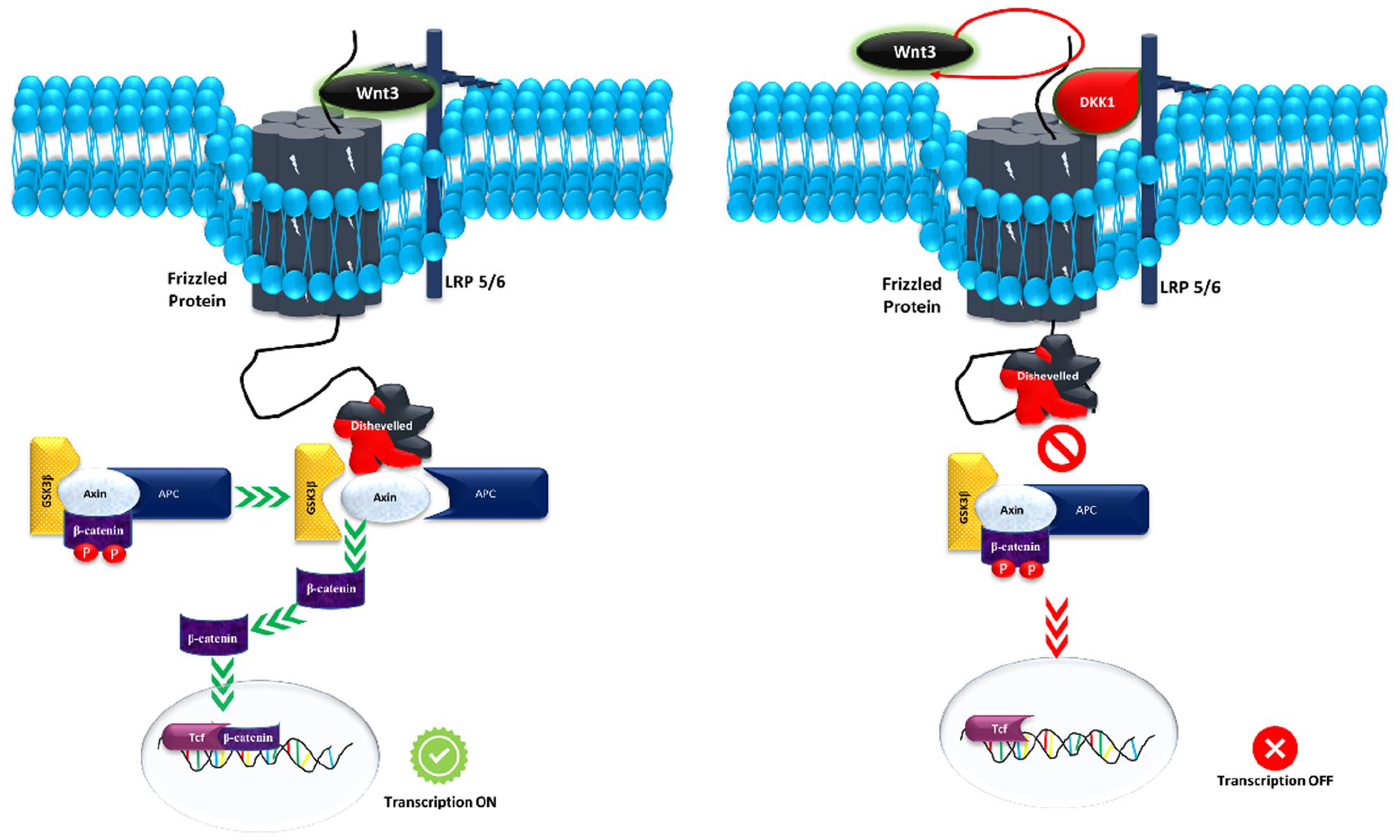
The schematic diagram represents the Wnt signaling pathway which is initiated when Wnt molecules bind to the classical frizzled receptor and the coreceptor LRP receptor, leading to the activation of Dishevelled protein (Dvl). Consequently, the “destruction complex” disassembles, causing the inactivation of GSK3β and the stabilization of β-catenin within the cytosol. This stabilized β-catenin is subsequently transported to the nucleus, where it facilitates the expression of target genes.
Therefore, if the interaction of DKK1/LRP6 is inhibited, it might be helpful for the positive regulation of Wnt signaling and subsequently, it will be useful to treat AD. 12 Till date, there is no Food and Drug Administration (FDA)–approved drug are available against DKK1-LRP6 interaction. The first identified inhibitor DKK1/LRP6 was NCI8642 (gallocyanine), 13 but it is a week inhibitor and not able to cross the blood-brain barrier (BBB). 14 - 16
There is a continuous need to identify new agents with a significant ADME (Absorption, Distribution, Metabolism, Excretion) profile and fewer side effects than currently prescribed AD treatment drugs. In this study, we used a structure-based virtual screening approach to accurately screen for novel inhibitors of the DKK1-LRP6 interaction, with the aim of developing potential therapeutics for AD.
The Binding Site in the Low-Density Lipoprotein Receptor–related Protein 5/6 Receptor for DKK1
The DKK1 protein is a secreted glycoprotein that specifically inhibits Wnt/β-catenin signaling by interacting with the extracellular domain of LRP5/6. The C-terminal of DKK1 (DKK1c) has a long loop with residues from 222 to 231 that are involved in the interaction with both copies of LRP6 propeller E3-E4 (interface A and B). These copies have a conserved hydrophobic patch (Trp850, Tyr 875, Phe836, Met 877, Trp767, Tyr706, Ile681). 7 The hydrophobic patch region (Phe836, Trp875, and Met877) of LRP5/6-E3 interacts with residues 226-231 of DKK1c. Other interactions are salt-bridge and Pi-Pi interactions between LRP6 (Asp830) and DKK1 (Arg191). 7 The 2 independent copies of LRP6E3-E4 protein interact with DKK1c in a sandwich and asymmetric manner. 9 The asymmetric unit is formed by the 2 LRP6 and 1 DKK1 molecule, in which the structure of LRP6 does not change in binding with DKK1 (Figure 2).

Structure of LRP6-DKK1 (as surface diagram: DKK1c) complex and hydrophobic interactions depicted by the blue rings in between of 2 copies of LRP6 (E3-E4) DKK1 C-terminal.
Materials and Methods
All computational processes were performed on Schrödinger software Maestro suite version 202017 and GROMACS Software 18 on an HP desktop with an operating system consisting of Linux Ubuntu OS18.04.02 LTS platform. Various modules such as LigPrep, Protein Preparation Wizard, Glide docking, Enrichment analysis for validating the virtual screening, Qik-Prop, molecular mechanics-generalized Born surface area (MM-GBSA) were used from Schrödinger software.19,20 Molecular dynamic (MD) simulations, binding free energy (BFE) calculation (molecular mechanics/Poisson-Boltzmann surface area [MM/PBSA]-based), and energy decomposition analysis (EDA) were performed using GROMACS. 18 The structure-based virtual screening approach was employed to identify the potential DKK1/LRP6 interaction inhibitor as summarized in Figure 3.

Flowchart of in silico study.
The Protein Selection, Validation, and Preparation
In structure-based molecular designing, the use of accurate protein structure is important. The negative regulation of Wnt signaling has been associated with AD. The 3-dimensional (3D) crystal structure of the LRP6-DKK1 protein was retrieved from Protein Data Bank (PDB ID:3S8V). 21 The selected crystal structure protein was first validated using of PROCHECK tool that is available on the SAVES v6.0 server. 22 It is a useful tool to predict stereochemical efficiency of selected protein. We can also examine the geometry of residues, quality of the selected protein structure via Ramachandran plots. This PROCHECK tool is used to assure the quality of protein structure for further docking purpose. The protein was prepared using the protein preparation wizard tool that involved pre-processing with the filling of missing loops and side chains, followed by optimization at physiological pH and minimization of constraint with Optimized Potentials for Liquid Simulations (OPLS) force field. 23
Compound Library Preparation
The CNS small molecules library with 54 513 compounds was downloaded from the Asinex database that is freely available. 24 The LigPrep tool was used to prepared the ligands molecules followed by geometric minimization using of force field (OPLS_2005) with retained specified chirality at pH 7.0 ± 2.0. The 2-dimensional (2D) structure was converted into 3D structure.
Structure-based Virtual Screening
The virtual screening is an applicable method to investigate the lead molecule for further research purposes against the targeted disease. Protein structure–based computational estimation of small molecule binding is a widely used approach by researchers involved in drug design studies. This method is helpful in screening out a million compounds from larger databases in a tandem process within a few hours. After the preparation of ligands, the grid was generated in the target crystal protein structure (3S8V:PDB ID) against the DKK1 binding site. The X, Y, Z co-ordinates were 14.51, 8.27, and 3.58. The size of the docking area was 10 Å was covered.
The glide module of Maestro software was used to conduct virtual screening, during which docking protocols were applied. Specifically, the high-throughput virtual screening (HTVS) mode was run with a weighting of 50%, followed by the standard precision (SP) mode with 30%, and finally, the extra precision (XP) mode with 10%, using a selected crystal structure (3S8V) and a prepared library of ligands. The molecules were selected on basis of higher docking score, ie, best fitting in the binding cleft. 19
Enrichment Analysis
Enrichment analysis was applied to validate the virtual screening process using Maestro suite (in task → receptor-based virtual screening → enrichment analysis). 25 The enrichment factor (Ef) reflects the number of actives identified throughout the virtual screening procedure. Conceptually, the Ef metric measures the how many actives we found within a defined “early recognition” fraction of the ordered list relative to a random distribution. 26 The Ef was calculated using the following formula

where Nexperimental is the number of experimentally found structures in the top x% of the sorted database, Nexpected is the number of expected active structures, and Nactive is the total number of active structures.
The ROC (Receiver Operating Characteristic) analysis is commonly used to evaluate the effectiveness of virtual screening methods. It represented as the equivalently by plotting the fractions of true-positive rate (TPR) vs the false-positive rate (FPR). The total 1000 decoy molecules were used for the validation of virtual screening.
Molecular mechanics-generalized Born surface area calculation
To determine the negative BFE of the ligand-protein complexes (docked structure), the Prime MM-GBSA method was used. The docked structures were inputted into the Prime MM-GBSA tool, which used the OPLS-3 force field and the VSGB solvent model to calculate the BFE. In addition, the flexibility of residues was limited to a distance of 5.0 Å. 27
Estimation of Absorption, Distribution, Metabolism, Excretion, and Toxicity Properties
Most of the lead molecules fail in clinical trials due to poor ADME prediction and is bigger a hurdle for novel drug development programs. The ability to recognized unsuitable candidates at an early stage may significantly decrease loss of time and money while also streamlining the whole development process. The ADME profile of compounds was determined using of Qik-Prop 20 on the basis of physicochemical and in silico pharmacokinetic descriptors. The molecular weight of selected ligands should be in less than 500 Da. The BBB permeability was analyzed of all ligands that have acceptable permeability value −3.0 to 1.2. The aqueous solubility (QPlogS) was also checked of ligands that shown the acceptable range −6.5 to 0.5. QPlogPo/w analysis was applied for understanding octanol/water partition coefficient which has acceptable range between −2.0 and 6.5 of all compounds. Caco-2 permeability was also analyzed to check gut blood barrier permeability. The qualitative prediction of human oral absorption with range 2 and 3 (1 = poor, 2 medium, 3 = higher). Molecules lying within the acceptable range of these properties, pertaining to showing good drug-likeliness, were selected for lead optimization study, as described in Tables 1 and 2.
The result of virtual screening process, MM-GBSA, MD simulation, and BFE calculation.

Abbreviations: BFE, binding free energy; MD, molecular dynamic; MM-GBSA, molecular mechanics-generalized Born surface area; RMSD, root mean square deviation.
The total active count and active percentage in N% of results.

Abbreviations: AUC, area under the curve; BEDROC, Boltzmann-enhanced discrimination of receiver operating curve; ROC, receiver operating characteristic.
Molecular Dynamic Simulation
The MD simulations were performed of the complex of DKK1 inhibitor with LRP6 receptor using GROMACS version 2021.04 through gmx_qk.28-29 Topology for the receptor was generated using pdb2gmx with a CHARM27 force field and TIP3P water model. Sodium and chloride ions were added to the box to neutralize the system. Energy is minimized for the solvated system within defined periodic boundary conditions (PBCs) and further equilibrated by employing NVT (constant number:N; constant-volume:V; and constant-temperature:T) and NPT (constant number:N; constant Pressure:P; and constant-temperature:T) at 50 000 nano steps. The last frame of the equilibrated system was submitted for the MD simulation for 100 ns. Post-MD simulation trajectories were analyzed using VMD after removing the PBCs and further calculated the C-alpha RMSD (Root Mean Square Deviation) using gmx rms module with the reference of first frame structure. 18
Principal component analysis
We can get precise information via principal component analysis (PCA) regarding each residue’s functional significance and residual motions through the C-alpha atomic co-ordinates. A covariance matrix comprises the atomic fluctuation of each residue’s C-alpha, providing us with orthogonal eigenvectors and eigenvalues. The eigenvalue gives the amplitude of the motions and eigenvectors specify the direction of the motions. The set of principle components (PCs) represented by these eigenvectors and related eigenvalues can be used to define the moments characteristics. Based on the first 2 principal components (orthogonal eigen vectors), structural fluctuation or variance of displacement during dynamic simulation was captured and plotted using bio3D R package. 30 It sampled the most of the structural displacement and provides more insights of conformational changes of our system during dynamic simulation run.
Dynamic cross-correlation map
The atomic motions (correlated and noncorrelated) of complex protein residues were analyzed using of Dynamic Cross-Correlation Map (DCCM) in bio3D web application. 30 To generate a covariance matrix, we simply took C-alpha atomic co-ordinates of each residue to reduce statistical noise. The following equation was used to calculate the covariance matrix element Cij

where Δri and Δrj represented the displacement from the average position of atoms (ith and jth) with the time. The time average for the entire trajectory is shown by the angular bracket. The value of cross correlated varies in between −1 and +1. The positive and negative value indicated the positive correlated motions and anti-correlated motion, respectively. Here, we have considered the final 90 ns production simulation trajectories for the analysis. 31
Binding Free Energy Calculation and Energy Decomposition Analysis
The model exhibiting least deviation in 100 ns classical MD simulation, trajectories (100 frames, removed PBCs) was subjected to the MM/PBSA-based BFE calculation using g_mmpbsa tool 29 . The equation for BFE calculation is as follows

where x is the ligand or the protein or ligand-protein complex, Gsolvation is the energy of solvation, EMM is the average molecular mechanics potential energy in vacuum, and TS is the configuration entropy (contribution of entropy temperature and S entropy). Energy decomposition analysis calculations were followed by the EDA. Energy decomposition analysis was performed to measure the contribution of each and every amino acid residue of the complex in the BFE. It was measured using g_mmpbsa tool with the python script MmPbSaDecomp.py
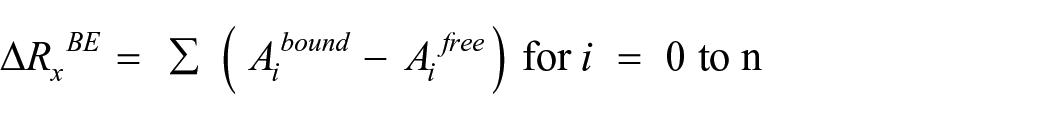
where Ai complex and Ai free are the energy of ith atom from x residue in bound and unbound forms, respectively, and n is the total number of atoms in the residue. Negative BFE plotted against a time frame to predict the complex stability.
Results and Discussion
Protein validation
The target LRP6-DKK1 protein-protein complex and the complete co-crystal structure were retrieved from the Protein Data Bank (PDB:3S8V). The stereochemical quality and 3D structure model of selected PDB:3S8V was evaluated by the PROCHECK tool and verified 3D tool which is provided by the SAVES v6.0 server. The 3D structure was verified 91.15% score of the residues has averaged 3D-1D score ⩾0.2, which considers at least 80% of the amino acid. The protein quality was assessed by the Ramachandran plot that was given a 78.8% score and 19.5% in the allowed region. The G factor was −0.05 with maximum deviation was 4.0, and the planar group is 100% with the limits (Figure 4).

Summary of the validation report for target protein structure by PROCHECK server.
Structure-based Virtual Screening and Molecular Mechanics-Generalized Born Surface Area Calculation
In this study, first grid was generated against the DKK1 binding site in the LRP6 dimer protein having XYZ co-ordinate center size is 14.51 × 8.27 × 3.58 as per previous studies. 32 Therefore, selected amino acid residues of both LRP6 interface A and B that involved in interaction with DKK1 are Phe836, Trp850, Asp874, Ser851, Trp767, Tyr875, Met877, Tyr706, and Ile681.7,32 Grid generation was followed by virtual screening of prepared library compounds against the DKK1-LRP6 binding site as shown in Figure 5. A total 149 compound were screen out. Furthermore, we selected top 6 molecules on the basis of highest docking score. The 6 compounds were shown significant negative docking scores in the comparison with control gallocyanine (a known DKK1 inhibitor) against the DKK1/LRP6E3E4 binding site. Ranked docking scores are ranged from maximum −8.276 of LAS 30169191 (Table 1). Furthermore, we performed MM-GBSA of all selected protein-ligand complex structure. We found all complexes were shown significant binding energy score. LAS 32378789 were shown the highest MM-GBSA score.

Represented the grid generation site in LRP6 protein and screened molecules on binding site.
Top ranked 6 ligands and a reference ligand (gallocyanine) were subjected to further evaluation in MD and BFE calculations.
Enrichment Analysis of Virtual Screening
The enrichment analysis was performed to evaluate the virtual screening protocol using of ROC and Boltzmann-Enhanced Discrimination of Receiver Operating Curve (BEDROC). The XP docking protocol was shown a significant area under the curve (AUC) of 0.94 and ROC of 0.91. The range of BEDROC was 0.309 (alpha = 160.9) as Table 2 shows. The percentage of active ligand was found more than 80%, and ROC curve was given in Figure 6A and B. The virtual screening process is verified by enrichment analysis.
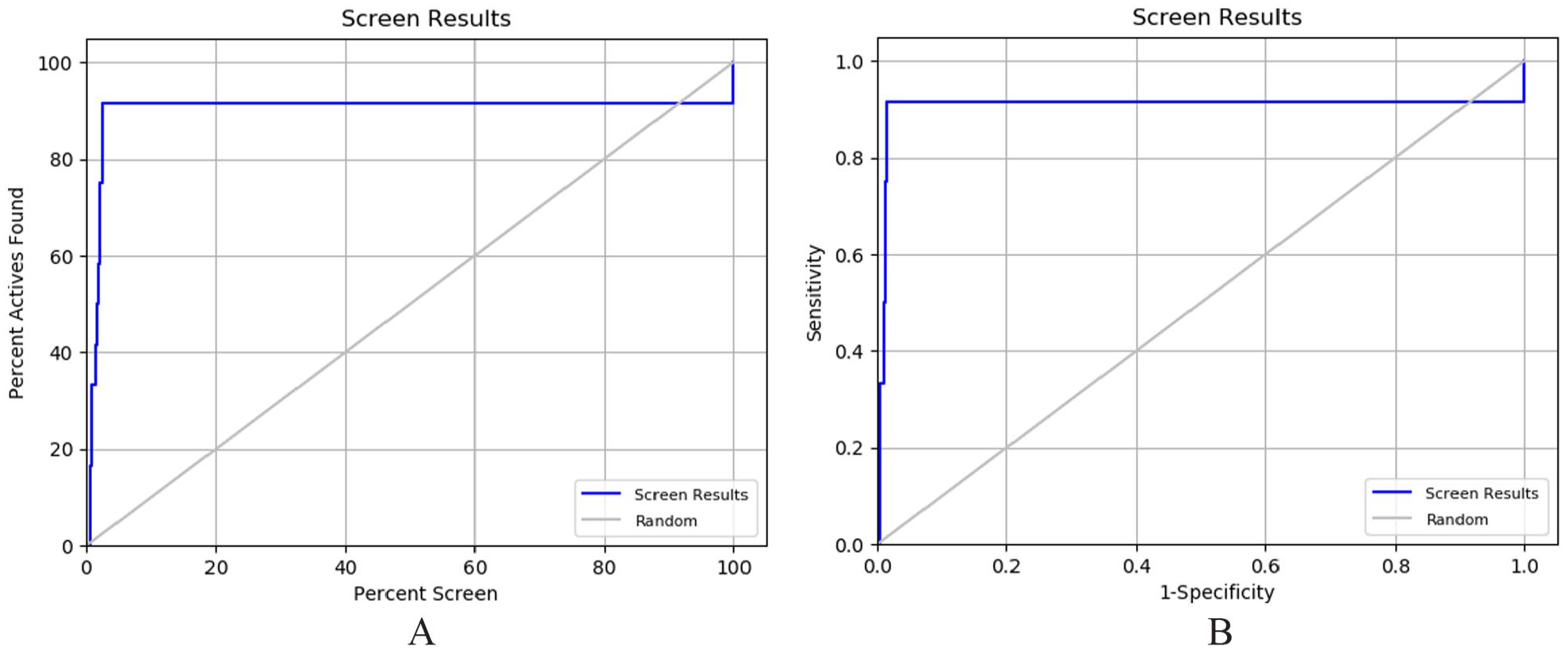
Represented the screen plot (A) and ROC plot (B) of virtual screening process.
Absorption, Distribution, Metabolism, and Excretion Prediction
Identifying a therapeutic compound for AD not only depends on targeted protein binding specificity but also depends on pharmacokinetics properties where BBB permeability is desired property. The ADMET prediction of selected 6 (+1 control) ligands was analyzed using of Qik-Prop module in the Schrödinger suite. The molecular weight of selected ligand was in between 268.361 and 323.437 Da. The BBB permeability was analyzed of all ligands that have acceptable permeability value −3.0 to 1.2. Aqueous solubility (QPlogS) was also checked of all ligands which shown all are in acceptable range −6.5 to 0.5. QPlogPo/w analysis was used for understanding octanol/water partition coefficient that has acceptable range between −2.0 and 6.5 of all compounds. Caco-2 permeability was also analyzed to check gut blood barrier permeability only found in LAS 29757582 compound, whereas all compounds were shown higher range from 500. The qualitative prediction of human oral absorption was found in all selected ligands with range 2 and 3 (1 = poor, 2 medium, 3 = higher). All of the ligands were in the acceptable range of pharmacokinetics properties. Therefore, all were further included for MD simulation with receptors (Table 3).
The pharmacokinetic profile of docked molecules based on ADME prediction.

Abbreviation: MW, Molecular weight; QPlogPo/w, octanol/water partition coefficient; QPlogS, aqueous solubility; Qppcaco, Caco-2 cell permeability in nm/sec; Qplogbb, brain/blood partition coefficient; HOA, Human oral absorption.
Molecular Dynamic Simulation
Virtual screening and ADMET profile prediction were followed by classical MD simulation analysis. The top ranked 6 (+1 control) ligands based on docking score and a reference ligand (gallocyanine) were subjected to further evaluation in MD simulations. Therefore, we further performed an MD simulation study of all complexes (protein-dimer-ligand) and apo form (without ligand) of LRP6 protein dimer structure at 100 ns. The MD data from GROMACS provide valuable insights into the LRP6 chains A and B intermolecular interaction. The LRP6 (apo) dynamics simulation analysis suggested that there is a very unstable association between 2 LRP6 chains and varies at average RMSD was 1.741 nm at 100 ns. Now, as the inhibitory active site lies at the interface of 2 LRP6-dimer receptors and a relatedly lower frequency of stable complex formation at that site. Hence, in this study, the features of LRP6 apo form have been taken as a ground reference point. Among all complexes, LAS 29757942 and gallocyanine were shown very high RMSD values 1.56 and 1.376 nm, respectively. Another side, LAS 32378789 and LAS 29757582 were shown lower RMSD values 0.542 and 0.717 nm (stabilized complex).
The RMSD values ascending order was LAS 32378789 < LAS 29757582 < LAS 29984441 < LAS 30169191 < LAS 32378790 < gallocyanine < LAS 29757942 (Figure 7 and Table 1).

C-alpha RMSD (nm) during the course of long run (100 ns) MD simulations for different protein-dimer-ligands.
Principal Component Analysis
Principal component analysis was employed to identify the significant global motions or statistical expression conformations that were observed during the 100 ns trajectory. 33 Principal component analysis was performed on the MD simulation trajectories of LRP6 in its apo form, as well as when bound to LAS 29757582, LAS 29757942, and LAS 29984441 molecules. The purpose of the analysis was to interpret the random global motions of the amino acid residues’ atoms (Figure 8). First 3 PCs (35.7%, 27.3%, 13.4%; cumulative variance explained 76.4%) explained the most of the structural conformations attained during dynamic simulation from blue to red convergence in case of LAS 29757582 as compared with the apo form. Other one LAS 29984441-LRP6 bound form was shown the structural conformation from blue to read as seen in PC1 (48.9%), PC2 (20.6%), and PC3 (11.1%), and cumulative variance explained was 80.6%.

PCA result of LRP6 trajectory with instantaneous conformation (trajectory frames), color from blue to red in order of time (convergence). for PC1, PC2 and PC3 with eigen vectors vs proportion of variance explained. (A) Apo form (protein only), (B) LAS 29757582, (C) LAS 29757942, and (D) LAS 29984441.
Low-density lipoprotein receptor–related protein 6 and LAS 29757942 complex was similar as apo form conformations stated using PC1 (76.8%) and PC2 (15.6%); however, PC2 and PC3 (3%) explain structural conformation displacements as compared with the apo form. It is feasible to distinguish the primary motions present in the trajectory, as well as the crucial motions needed to bring about conformational changes.
Dynamical Cross-Correlation Map
An R package bio3D was used to analyze the structure, conformational heterogeneity, and residual fluctuations of target protein families. The positive and negative correlation movement of amino acid residues was depicted by cross-correlation graph. The pairwise-correlated graph was created between the function of residue indices i and j. Residue cross-correlations explained the conformational displacement in the presence of different ligands (LAS 29757582, LAS 29757942, LAS 29984441) as compared with the apo form plotted in Figure 9. Majorly positive correlation found 1 to 250 residues which were reverted in the case of ligand LAS 29757582 and LAS 29984441, and in addition, all the correlations were reversed as depicted in Figure 7. Similar pattern of movements correlation was found in the case of LAS 29757942 and but lesser than LAS 29757582 and LAS 29984441 and similar to the apo form.

Residues cross-correlation heat map: (A) apo form, (B) LAS 29757582, (C) LAS 29757942, and (D) LAS 29984441 (cyan-pink +1 to −1 correlation coefficients).
Binding Free Energy Calculation and Energy Decomposition Analysis Results
After the MD simulation, thermodynamic-based MM/PBSA was calculated for every 10th trajectory frame (total frames = 1000) to predict the stability of ligand binding with selected protein pocket. The maximum negative binding energy was found at −578.045 ± 5.142 kJ/mol, −531.055 ± 3.325 kJ/mol, and −476.685 ± 7.370 kJ/mol of the LAS 29757582, LAS 29984441, and LAS 29757942 with LRP6 protein, respectively (Figure 10 and Table 1). The gallocyanine that was taken into account as a standard control resulted positive values of BFE. Hence, the ligands showing positive energy values were excluded from EDA and further analysis. The line plot of negative BFE revealed the clear energy minima during the long course of MD simulation runs. Respective energy minima conformation attained is illustrated in Figure 11. Most of the leads interacted with DKK1 binding site residues of chains A and B of LRP6. Residues interacting at binding site in complex formation were Tyr875 (chain B) and His834, Tyr875, and Trp850 (chain A).

The binding free energy change (kJ/mol) during the course of long run (100 ns) MD simulations trajectories for different protein-dimer-ligands.
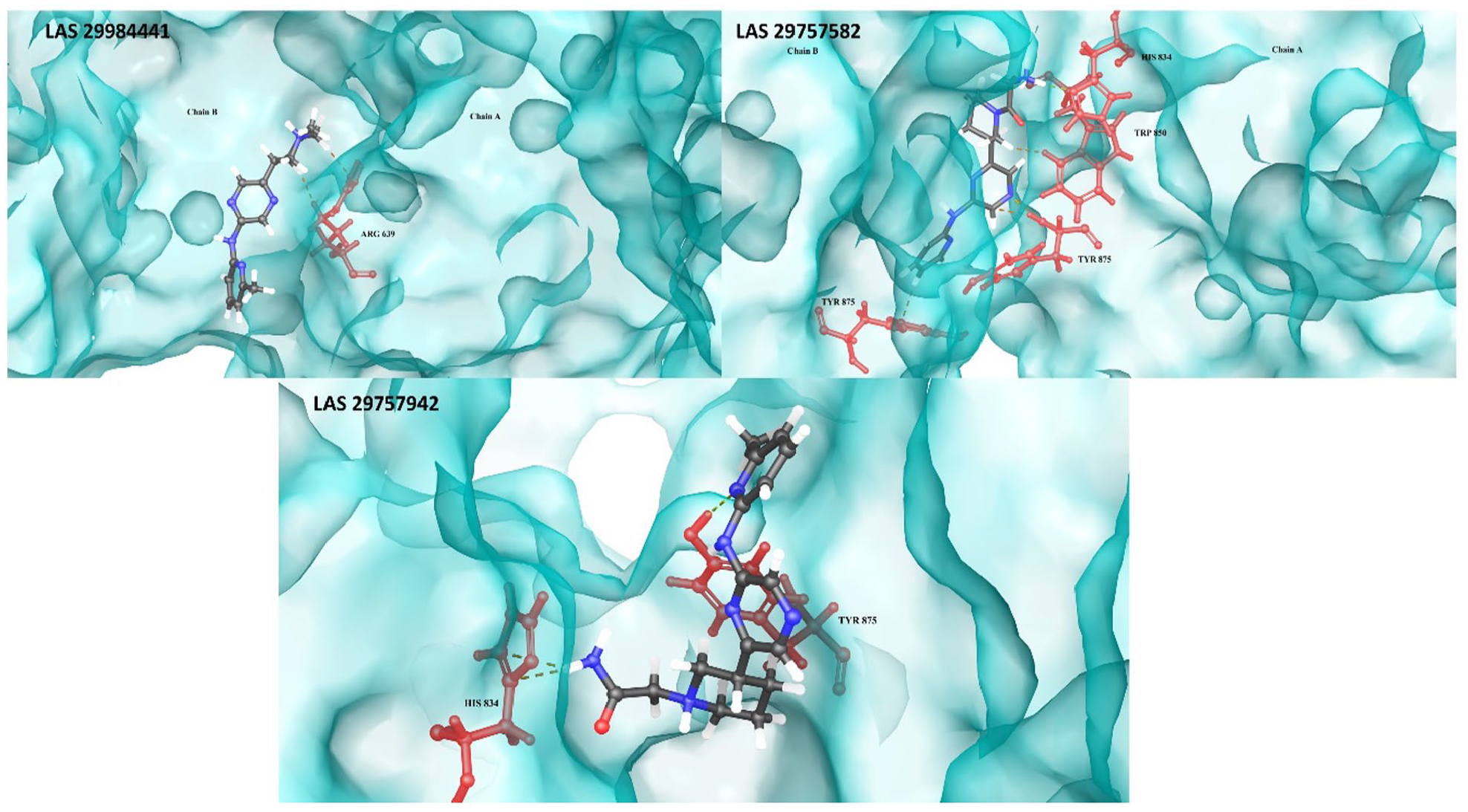
The protein-dimer-ligand interaction profile for stable complex formation (dashed line representing interactions between ligand (gray) and the respective target site residues (red)).
In detail, LAS 29757582-LRP6 complex formation stabilized by the interactions with both chains A and B of LRP6 which includes H-bonds between N-terminus of Tyr875 (chain B) and N-terminus of A chain residues including His834, Tyr875, and Trp850. The protein-ligand interactions were supported by the EDA results (Supplementary Table S1) as per most negative binding energy contributed by the exact same residues as showing in surface diagram of LAS 29757582-LRP6 complex. Second-ranked stabilized LAS 29757942-LRP6 complex also consists similar H-bond interactions between Tyr875 of chain A only. The addition of non-covalent interactions, i.e., ionic interactions with His834 (B chain), plays a vital role in the stability of complex. Third-ranked stabilized LAS 29984441-LRP6 complex involves only ionic interactions with the binding site residue ARG639 of chain A.
The receptor-ligand interactions were supported by the EDA. All complexes (protein and ligand) were subjected to EDA, and residue viz contribution to the total negative BFE was evaluated. Energy decomposition analysis elucidated binding site residue interaction profiling for selected LRP6 (A and B interface) and leads, ie, LAS 29757582-LRP6, LAS 29757942-LRP6, and LAS 29984441-LRP6 interaction, toward stabilizing the complex formation as per Supplementary Table S1. Hence, the exact same residues Tyr875, His834, Trp850, and Arg639 contributed in most negative BFE as discussed above.
Resulted 3 lead compounds (LAS 29757582, LAS 29757942, and LAS 29984441) were belonging to SL#044 CNS set of Asinex library. These molecules were designed using in vitro screening of Asinex’s BioDesign molecules have exposed a number of CNS-like molecules with high PAMPA-BBB permeability properties (Pe > 10 × 10− 6 cm/s).34,35 Such properties are primary requisite for therapeutics for neurological disorders, such as AD. However, to determine their efficacy and safety, the molecules need to undergo wet lab validation following computational screening. Therefore, further studies are necessary to evaluate their effectiveness in animal disease models of AD.
Conclusions
In this study, we developed a structural-based virtual screening in silico algorithm to identify a new chemical entity that could target the interaction of LRP6-DKK1 protein in AD. Using virtual screening, we identified 6 leads that showed significant binding interactions. We then applied ADMET filters and conducted MD simulations and MM/PBSA-based negative BFE calculations. Our extensive computational analysis resulted in the identification of 3 potential hits—LAS 29757582, LAS 29984441, and LAS 29757942—that could block the interaction of DKK1 with LRP6 (A and B interface) protein. These results were supported by negative BFE calculations. It is important to note that the antagonistic/inhibitory effects of these leads will need to be validated in wet lab experiments, which is a limitation of this study.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322231183762 – Supplemental material for A Novel Inhibitor of DKK1/LRP6 Interactions Against the Alzheimer Disease: An Insilco Approach
Supplemental material, sj-docx-1-bbi-10.1177_11779322231183762 for A Novel Inhibitor of DKK1/LRP6 Interactions Against the Alzheimer Disease: An Insilco Approach by Manisha Prajapat, Harvinder Singh, Gajendra Chaudhary, Phulen Sarma, Gurjeet Kaur, Ajay Prakash Patel and Bikash Medhi in Bioinformatics and Biology Insights
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
MP, HS: Conceptualization, Methodology, Writing- Original draft preparation, Data curation. GC: Figures and table preparation, PS and GK: Software, Investigation. AP and BM : Supervision, Validation, Reviewing, Editing and finalization of draft.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.