
Abstract
The rapid and global spread of the novel coronavirus severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) has raised serious public health concerns, including in Mauritania. We sequenced and analyzed the entire genome of 13 SARS-CoV-2 virus strains isolated from polymerase chain reaction (PCR)-positive symptomatic patients sampled from March 3 to May 31, 2021 to better understand SARS-CoV-2 introduction, propagation, and evolution in Mauritania. A phylogenetic tree using available data from the EpiCoV GISAID database and a variant network with non-Mauritanian sequences were constructed. Variant analysis of the 13 Mauritanian SARS-CoV-2 genome sequences indicated an average mutational percentage of 0.39, which is similar to that in other countries. Phylogenetic analysis revealed multiple spatiotemporal introductions, mainly from Europe (France, Belgium) and Africa (Senegal, Côte d’Ivoire), which also provided evidence of early community transmission. A total of 2 unique mutations, namely, NSP6_Q208K and NSP15_S273T, were detected in the NSP6 and NSP15 genes, respectively, confirming the aforementioned introduction of SARS-CoV-2 in Mauritania. These findings highlight the relevance of continuous genomic monitoring strategies for understanding virus transmission dynamics and acquiring knowledge to address forthcoming sources of infection in Africa.
Keywords
Introduction
There have been multiple incidents of respiratory problems reported in Wuhan, Hubei Province, China, following the viral outbreak in December 2019. 1 Coronavirus disease 2019 (COVID-19) was later revealed to be induced by a novel coronavirus,2,3 which was thereafter labeled severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). 4 At the outset of the epidemic in China, human-to-human transmission occurred mainly between relatives and friends who had close contact with patients or carriers;5-7 afterward, community transmission was observed.
As the primary infection site, SARS-CoV-2 actively spreads in the lungs. This active proliferation causes a flood of inflammatory cytokines, which, if not reduced, contribute to the worsening of the disease pathology.8,9 The ongoing SARS-CoV-2 outbreak was declared a pandemic by the World Health Organization (WHO) on March 12, 2020. 10 As of March 7, 2023, there had been 759,408,703 reported cases worldwide, with 6,866,434 deaths (John Hopkins Center [JHC]), 11 whereas Mauritania has declared 63,439 confirmed cases and 997 deaths. On March 13, 2020, the first case of COVID-19 in Mauritania was reported in the country’s capital, Nouakchott, from an individual who had traveled to Italy. In less than 2 weeks (March 15–29, 2020), there was an alarming increase in the number of positive polymerase chain reaction (PCR) tests as the number of cases increased exponentially. A state of health emergency has been declared to combat the spread of the virus.
The availability of vaccines and the strategy developed by the Ministry of Health to combat the pandemic has shown a distinct drop in the number of positive cases detected per day. As of April 11, 2023, the number of vaccinated individuals has reached 447.82 million who have taken the first dose, 5.11 billion who have taken the second dose, and 2.74 billion who have taken the booster dose. 12
The continuing emergence of new variants in the world highlights the need to strengthen local sequencing capacity and genomic surveillance in low-setting regions for coordinated national and regional responses to SARS-CoV-2 infection and pandemics. However, there are still limited data on the genomic sequence of SARS-CoV-2 variants circulating in such countries, including Mauritania, a country on the Atlantic coast of Africa forming a geographic and social bridge between North Africa and the westernmost component of sub-Saharan Africa. This study aims to detect mutation profiles and phylogenetic lineages of circulating SARS-CoV-2 variants in Mauritania as a representative country of that region. Our analysis provided genomic evidence for the spread of different lineages of SARS-CoV-2, including AY.34.1, B.1.525 (initially detected in Nigeria and the United States, and carrying the E484K mutation and 2 deletions in the spike protein found in the 501Y.V1 variant), B.1.620, and B.1.1.318 (with the E484K mutation and a substitution in position 681 of the spike protein), in Mauritania. This is the first genomic investigation of the SARS-CoV-2 virus in this country, which will shed light on the patterns of infections that drive the epidemic in the North Sub-Saharan Africa transitional region. We performed variant analysis and genome-wide phylogeny of 13 SARS-CoV-2 strains to better understand the molecular epidemiology of the COVID-19 outbreak in Mauritania. All samples were isolated from Mauritanian patients in Nouakchott from March 3 to May 31, 2021 (Table 1).
Metadata of SARS-CoV-2 strains analyzed in this study.

Abbreviation: SARS-CoV-2, severe acute respiratory syndrome coronavirus-2.
Materials and Methods
Sample collection and processing
The Mauritanian-related SARS-CoV-2 genomes used in this study were sequenced on an Ion Torrent GeneStudio S5 Sequencer (Thermo Fisher Scientific, Waltham, MA, USA) at The National Institute of Public Health Research laboratory (INRSP, Mauritania) and deposited in the GISAID database (https://www.gisaid.org/) (Table 1).
Variant analysis, gene alignment, and annotation of the SARS-CoV-2 genomes
Variant analysis of the 13 Mauritanian SARS-CoV-2 sequences was performed using CoVsurver (https://www.gisaid.org/epiflu-applications/covsurver-mutations-app/) by mapping them to the Wuhan SARS-CoV-2 sequence reference (WIV04).
Phylogenomic analysis
To find closely related sequences, we used AudacityInstant, and the resulting sequences were aligned with the Mauritanian sequences using Mafft v2.10.0.13,14 We used Modeltest v0.1.7 15 to find the best-fits maximum likelihood model and then fed it to iqtree v2.2.0.3 16 to construct it. ITols v4.3.3.5 17 was used to visualize the phylogenetic tree.
Results and Discussion
Phylogenetic analysis of 13 Mauritanian SARS-CoV-2 genomes
The complete genomes of SARS-CoV-2 viruses from 13 Mauritanian patients were sequenced and mapped against the EpiCoV Gisaid database, which contained approximately 9.8 million genomes (April 2022). Using AudacityInstant, we found a total of 1017 SARS-CoV-2-related genomes from different countries. It thus allowed us to search the entire EpiCoV database for closely related sequences, providing valuable metadata about each related sequence, such as clade, lineage, location, variant, and collection date (Figure 1).

Overview of the closest related SARS-CoV-2 genomes. (A) The different countries that are closely related to the most prevalent SARS-CoV-2 sequences, including the Mauritanian sequences, the SARS-CoV-2 strains identical to the Mauritanian strains, and the different lineages of the most frequent mutations detected in these sequences similar to the Mauritanian strain, which confirms that the Mauritanian strain comes from different origins. (B
The expanded family tree is divided into 7 clades that accordingly correspond to the major SARS-CoV-2 strain types GR, GRY, GH, GK, G, S, V, L, and O. The 3 strain types G, GR, and GH are distributed worldwide. They first appeared in February 2020, according to GISAID, where the G strain is the original clade from which both the GH and GR strains descended. 18 The analysis showed that the Mauritanian isolates were grouped into 4 independent clades. The sequence Sample ID: S1_NKC_04_08 (Figure 2) clusters with sequences from the USA GISAID ID EPI_ISL_4965219 in the GR clade.

Related genomes of Mauritania sequence S1_NKC_04_08.
The Mauritanian sequences (Sample IDs: S2, S3, S4, S5, S12, S13) (Figures 2 to 5, 12, and 13), which are grouped together in the GK clade, are similar to those from Switzerland/Zurich (GISAID ID: EPI_ISL_3507737), the United States (GISAID ID: EPI_ISL_7236215), Belgium/Antwerpen (GISAID ID: EPI_ISL_8202826), and France (GISAID ID: EPI_ISL_3477828) (Figures 6 to 8).

Related genomes of Mauritania sequence S2_NKC_04_08.

Related genomes of Mauritania Sequence S3_NKC_04_08.
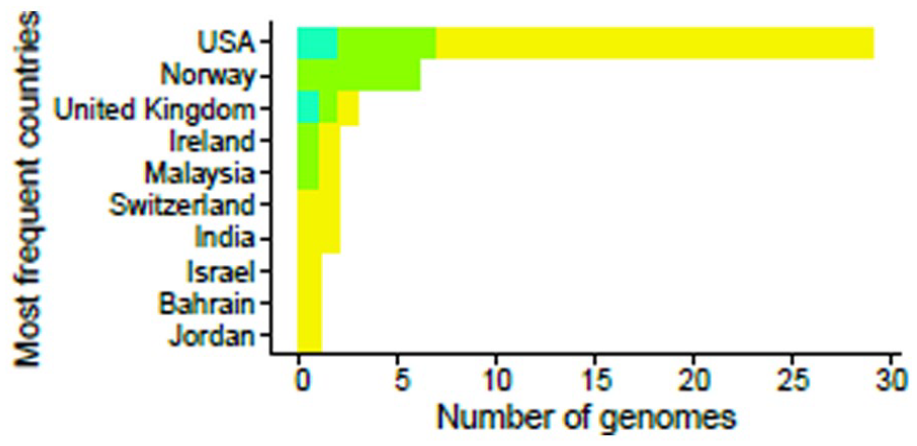
Related genomes of Mauritania sequence S4_NKC_04_08.

Related genomes of Mauritania Sequence S5_NKC_04_08.

Related genomes of Mauritania sequence S12_NKC_25_08.

Related genomes of Mauritania Sequence S13_NKC_25_08.
In the G-type strain, 4 Mauritanian sequences are clustered together (Sample IDs: S6, S7, S8, S9, S11) (Figures 9 to 13) with other Mauritanian sequences not included in this study (GISAID ID: EPI_ISL_11380685, EPI_ISL_11033244, EPI_ISL_11380682), Senegal/Dakar (GISAID ID: EPI_ISL_8528458), and Cote d’Ivoire/Bouake (GISAID ID: EPI_ISL_3545640) sequences.

Related genomes of Mauritania sequence S6_NKC_03_08.

Related genomes of Mauritania Sequence S7_NKC_03_08.

Related genomes of Mauritania sequence S8_NKC_03_08.

Related genomes of Mauritania Sequence S9_NKC_25_08.

Related genomes of Mauritania sequence S11_NKC_25_08.
Another sequence isolated from a patient in Mauritania (Sample ID: S10) (Figure 14) was grouped with a sequence from Senegal/Dakar (GISAID ID: EPI_ISL_8528001) in the GRY clade.

Related genomes of Mauritanian sequence S10_NKC_25_08.
Finally, a comparison of the Mauritanian sequence collected on May 21, 2020 (Sample ID: S6) (Figure 9) and the sequence collected on February 28, 2021 (GISAID ID: EPI_ISL_11380685) showed that the strains spread in Mauritania did not undergo major mutations, also reconfirming the spread of the virus by local community infection (Extended data, Phylogenetics Tree). 19 Because it was deposited after this work was completed, this sequence was not included in the overall analysis.
According to phylogenetic analysis, the viral outbreak in Mauritania was most likely the consequence of various introductions. We had several independent Mauritanian presentations, mainly from Europe and Africa. Robust statistical inferences are hampered by the limited number of genomes; nonetheless, the findings of this study provide an important understanding of the dynamics of the early introductions and local transmission of SARS-CoV-2 in Mauritania.
Variant analysis
The analysis of alterations that occurred at the amino acid level showed different mutations (Figure 15), of which 0.39% were known, whereas 2 unique mutations were found (Table 2): (1) F “NSP6_Q208K” mutation, representing a 73-nucleotide gap from the “G” clade, ie, the D614G protein spike variant, suggesting increased human host infectivity and virus transmission efficiency 20 and (2) “NSP15_S273T” mutation, consisting of a 3-nucleotide insertion with a gap of 13 nucleotides from the “GK” clade. NSP15, a coronavirus-specific uridine-specific endoribonuclease, is processed to avoid detection by host defense mechanisms. 21 Recent research suggests that, rather than being involved in viral RNA synthesis, NSP15 nuclease activity is important in evading host immune responses.22-25 Yuan et al 26 found the same unique mutations that characterized the Mauritanian samples, but the positions of the mutations were different.
List of different alterations at the amino acid level of SARS-CoV-2 genome sequences isolated in Mauritania.

Abbreviations: GISAID, Global Initiative on Sharing Avian Influenza Data; SARS-CoV-2, severe acute respiratory syndrome coronavirus-2; WIV04, Wuhan SARS-CoV-2 sequence reference.
Effect of Amino Acid Changes on the Spike Protein
Angiotensin-converting enzyme 2 (ACE2) is a functional receptor for coronaviruses through interaction with the virus spike protein and has been defined as a membrane-bound aminopeptidase.27- 29 We evaluated the effect of amino acid changes in the newly detected mutations (NSP6_Q208K and NSP15_S273T) in the SARS-CoV-2 genomes of strains collected in Mauritania on spike protein conformation and the impact of these mutations on the virus-host relationship (Figure 15).

The 3-dimensional structure of the SARS-CoV-2 spike glycoprotein in its interaction with human ACE2 reveals key amino acid substitutions. The green section depicts the ACE2 receptor and how changes in the spike protein affect the binding affinity between the SARS-CoV-2 spike receptor binding domain (RBD) and human ACE2. The structure displays a list of variations (nearest residue if in the loop/termini region). The figure was created employing the CoVsurver app from Gisaid.org.
Conclusions
Multiple spatiotemporal introductions of SARS-CoV-2 in Mauritania were revealed employing phylogenetic analysis and variant network analysis of SARS-CoV-2 genome sequences isolated from Mauritanian COVID-19 patients, most of which originated from Europe and Africa. These findings also showed that early community transmission occurred. A total of 6 lineages have been reported to be present in Mauritania, and 2 unique mutations have been detected: one in the NSP6 gene suggesting significantly increased human host infectivity and virus transmission efficiency and one in the NSP15 gene involved in the process of avoiding detection by host defense mechanisms. However, due to the absence of demographic and clinical data on the sequences of the majority of Mauritanian isolates, we were unable to draw possible associations between mutations and the clinical effects of the strains, whereas the lack of access to a large number of local isolates at the moment of this analysis hinders robust statistical conclusions. Nonetheless, the findings of this study provide intriguing insights into how the virus was initiated in Mauritania, as well as foundational knowledge for comprehending the dynamics of the virus’s early establishment and community transmission in Mauritania.
Footnotes
Acknowledgements
The authors acknowledge the INRSP laboratory of the National Institute of Public Health for depositing Mauritanian-related sequences used in this study in the GISAID database (https://www.gisaid.org/) (![]() ).
).
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: H.G. is a US NIH grant recipient through the H3abionet/H3africa consortium U24HG006941.
Declaration Of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
A. Abdelmalick: Data Curation, Formal Analysis, Investigation, S. Sehli: Visualization, Investigation, Writing – Original Draft Preparation, A. Idrissi Azami: Data Curation, Formal Analysis, Methodology, Software, N. Habib: Review & Editing, N. Al Idrissi: Review & Editing, Investigation, Resources, L. Belyamani: Review & Editing, Investigation, Resources, A. Houmeida: Review & Editing, Investigation, Resources, H. Ghazal: Conceptualization, Funding Acquisition, Investigation, Project Administration, Supervision, Review & Editing.