
Abstract
MicroRNAs (miRNAs) are single-stranded, endogenous, non-coding RNAs of 20–24 nucleotides that play a significant role in post-transcriptional gene regulation. Various conserved and novel miRNAs have been characterized, especially from the plant species whose genomes were well-characterized; however, information on miRNA in economically important plants such as pea (Pisum sativum L.) is limited. In this study, I have identified conserved and novel miRNA in garden pea plant leaves samples along with their targets by analyzing the next generation sequencing (NGS) data. The raw data obtained from NGS were processed and 1.38 million high-quality non-redundant reads were retained for analysis, this tremendous quantity of reads indicates a large and diverse small RNA population in pea leaves. After analyzing the deep sequencing data, 255 conserved and 11 novel miRNAs were identified in the garden pea leaves sample. Utilizing psRNATarget tool, the miRNA targets of conserved and novel miRNA were predicted. Further, the functional annotation of the miRNA targets were performed using blast2Go software and the target gene products were predicted. The miRNA target gene products along with GO_ID (Gene Ontology Identifier) were categorized into biological processes, cellular components, and molecular functions. The information obtained from this study will provide genomic resources that will help in understanding miRNA-mediated post-transcriptional gene regulation in garden peas.
Introduction
The garden pea is an economically important cool-season crop, which is grown across the globe. It is a rich source of protein, fiber, starch, phytochemical substances, and trace elements. Besides human consumption, pea has been grown as forage crops for cattle as well as a cover crop to minimize soil erosion (http://dpd.gov.in/Pea.pdf).
Due to the increase in population and consumer awareness about the consumption of healthy plant-based proteins, the demand for peas has been increasing in developed and developing countries. In terms of production, the pea is the fourth most important legume crop grown throughout the world. From 2022 to 2027, the pea market is expected to register a compound annual growth rate of 4.3%. (https://www.mordorintelligence.com/industry-reports/peas-market)
This protein-rich pulse crop is widely grown in China, India, the USA, France, Egypt, the UK, Pakistan, Algeria, Peru, and Turkey. India is the second largest producer of green peas in the world with a total production of 4.8 million tons (https://worldmapper.org/maps/green-peas-production/).
In plant species, microRNAs (miRNAs) and small interfering RNAs (siRNAs) are two important types of small regulatory RNAs. These two types of RNA differ in their function and biogenesis. In plants and animals, these small regulatory RNAs are highly conserved and regulate gene expression.
miRNAs are 20–24 nucleotides long non-coding RNAs that play a significant role in post-transcription gene regulation. It complements the target mRNAs and causes translational repression or target mRNA degradation. 1 In plants, RNA polymerase II transcribes miRNA genes into a primary transcript (pri-miRNAs). Ribonuclease III-like Dicer (DCL1) enzyme trims pri-miRNAs and produces miRNA precursors (pre-miRNAs) that had a stem-loop (hairpin) structure. Eventually, a short double-stranded RNA (dsRNA) is produced by the second cleavage of DCL1 on the stem-loop region of the hairpin. In the dsRNA, one strand acts as a mature miRNA. Subsequently, the processed miRNA incorporates with the RNA-induced silencing complex (RISC) and directs the RISC to the target complementary mRNA for translational inhibition or degradation.2,3 Plant miRNA involves in various biological processes such as signaling, 4 development, 5 and stress responses,6-11 and organ morphogenesis. 12
Formerly, miRNAs were identified using computational and hybridization-based experiments. As the computational-based method was entirely dependent on the availability of the EST or genome survey sequences in the database. If miRNA was expressed at a very low level, then it was difficult to detect miRNA using the hybridization-based method. On the contrary, using high-throughput next-generation sequencing (NGS) technologies like Illumina Solexa, Roche 454, and ABI SOLi, it is possible to differentiate lowly expressed miRNAs from well-characterized or poorly characterized plant species.13-18 Recent evidence indicates that plant miRNAs and siRNAs play a role in biotic and abiotic stress responses
The biotic and abiotic stress has been relegating the production of a garden pea. To minimize the damage caused by biotic and abiotic stress, plants regulate gene expression at transcriptional, post-transcriptional, and post-translational levels. A thorough understanding of the gene expression level at post-transcriptional will help in the development of new strategies that will enhance plant stress tolerance.
To understand the transcriptional response in garden pea plant, transcriptome analysis of pea tissues and organs were fully investigated. To gain insights into garden pea plant post-transcriptional and post-translational modification related to stress, there is a need to study miRNAs in various organs and tissues of pea plants under different conditions. As miRNAs can control various protein-coding genes associated with related gene families or genes that are involved in the same pathway, it is interesting to study miRNA-mediated post-transcriptional gene regulation in pea plants. 19
The current literature lacks research studies on miRNA related to pea plants. Therefore, in this study, to gain robust information about the entire repertoire of miRNA present in garden peas, a small RNA library was prepared from young garden pea leaves tissues, followed by deep sequencing and bioinformatic analyses.
Material and Methods
Sample preparation and extraction of total RNA
In this study, I have selected P. sativum (var. Arkel), which was easily available in the local market of India. The growth of this variety was vigorous, and the plant can grow up to 45 cm (http://agropedia.iitk.ac.in/content/vegetable-pea-varieties). Mature seeds of P. sativum (var. Arkel) were surface sterilized in 70% ethanol for 2 min and rinsed two times in sterile distilled water. The seeds were soaked in water overnight. Then, the seeds were sown in pots containing humus-rich soil. The plants were allowed to grow for a month in a net house. The plants were covered with plastic bags with holes. The young leaves were collected and immediately chilled in liquid nitrogen. The samples were stored at −80 C until RNA isolation. Utilizing the Trizol reagent, as per the manufacturer’s instructions, total RNA was extracted from the leaves.
Using Nanodrop Spectrophotometer, the RNA concentration and purity were estimated. A good quality RNA with a very low amount of protein contamination would have A260/A280 ratio greater than 1.8. Similarly, if the isolated RNA has a very less amount of polysaccharides contamination, then A260/A280 will be greater than 1.8. The total RNA extracted in this study has a concentration of 352.8 ng/ul, A260/A280 ratio of 2.19, A260/A230 ratio of 2.22 indicating a good quality RNA.20,21
Small RNA library preparation and deep sequencing
Small RNA sequencing (sRNA) library was prepared with TruSeq Small RNA Sample Preparation protocol (Illumina, San Diego, California, USA). 1000 ng of total RNA was used as starting material. 3' adapters were ligated to the specific 3’OH group of micro RNAs followed by ligation of 5' adapter.
Illumina Universal and Index Adapters utilized in this study were as follows:
5'-AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGA-3'
5'-CAAGCAGAAGACGGCATACGAGAT[INDEX]GTGACTGGAGTTCCTTGGCACCCGAGAATTCCA-3'
Reverse transcription of adapter-ligated fragments was performed using Superscript III reverse transcriptase (Invitrogen). The cDNA generated from the reverse transcription was enriched and barcoded by PCR amplification (15 cycles). Utilizing Polyacrylamide gel, the amplified library was size selected in the range of 140-160 bp followed by overnight gel elution and precipitation in the presence of glycogen, 3M sodium acetate (Sigma, Saint Louis, Missouri, USA), and absolute ethanol. Pellet was re-suspended in nuclease-free water (Invitrogen, Whitefield, Bangalore). The workflow of TruSeq Small RNA sample preparation is shown in Figure 1.
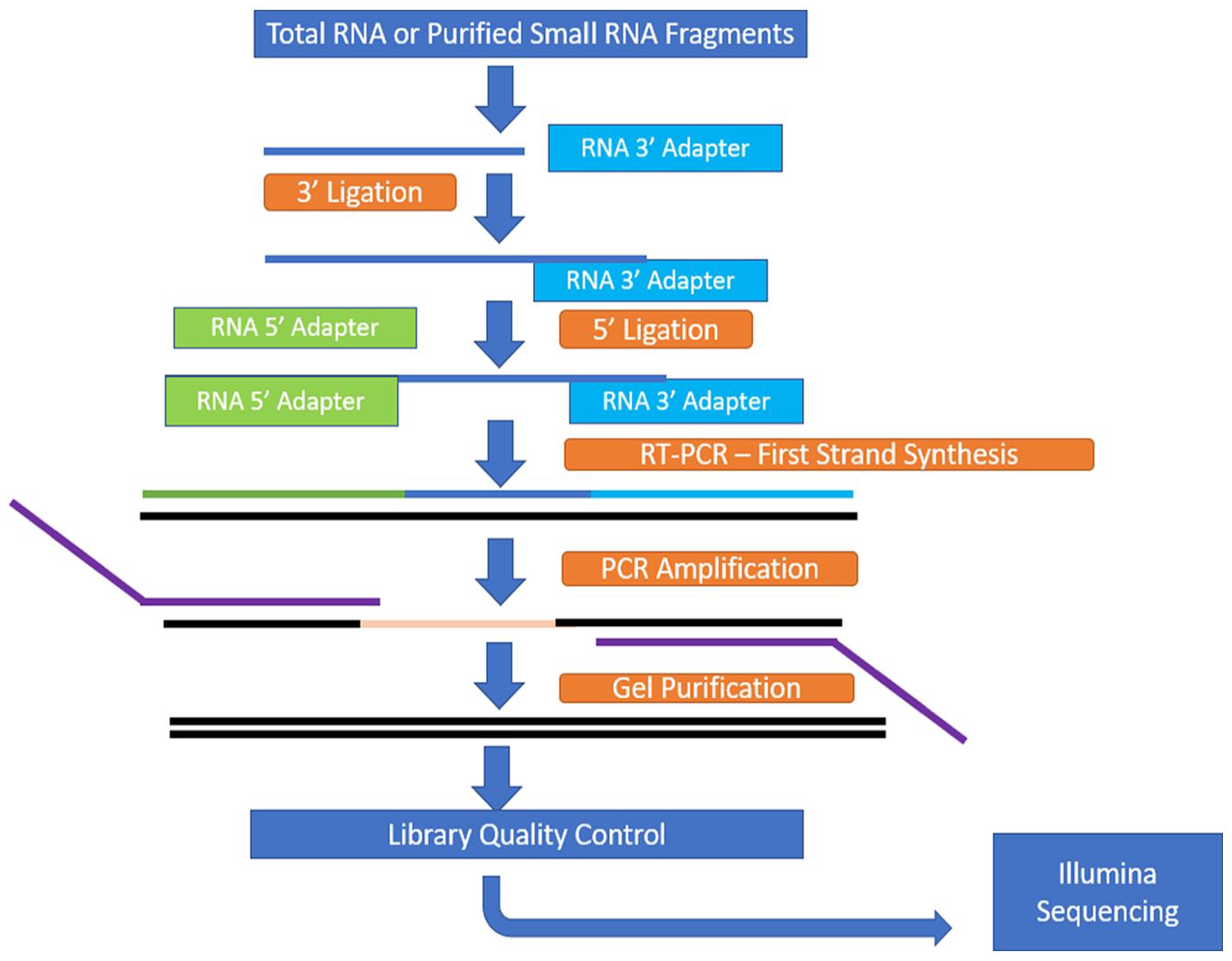
Overview of small RNA sample preparation.
Qubit fluorometer (Thermo Fisher Scientific, MA, USA) was used to quantify the Illumina-compatible sequencing library. The Qubit concentration of the small RNA library was 13.4 (ng/ul). Agilent 2100 Bioanalyzer was used to analyze the fragment size distribution. The fragment size of the Illumina-Compatible sequencing library ranges between 130 and 180 bp. Since the combined adapter size was approximately 120 bp, the effective user-defined insert size ranges between 10-60 bp. The raw NGS data of P. sativum young leaves sample was deposited in National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database with accession PRJNA882352.
Data analysis of small RNA transcriptome
After completion of the NGS run, the raw data were analyzed using UEA small RNA Workbench (Version 4.6; http://srna-workbench.cmp.uea.ac.uk).22,23 In Oracle VM VirtualBox, a virtual machine (VM) was created, and Ubuntu OS was installed on it. The adaptor sequences and low-quality reads were filtered and the sequences between 16 to 40 nucleotides were retained for further analysis. Then, using Rfam database, the reads that match with other ncRNAs such as rRNA, tRNA, snRNA, and snoRNAs were removed. 24 Glycine max genome was used as a reference genome. Using Bowtie software, the small RNA was aligned to the reference genome.25-27 The mapped and unmapped reads were separated using SAMtools. Using the CD-HIT program, a read count was generated.28-30 The mature sequences of miRNA were downloaded from miRbase (https://www.mirbase.org/), a local miRNA database was constructed, then a homology search was performed using the ncbi-blast-2.13.0 + program to identify the conserved miRNA with E-value of 0.001 and non-gap alignment.31-35 Subsequently, the unaligned reads were used for predicting novel miRNAs using the MIREAP program. A total of 29 novel miRNAs were predicted using the software; however, only 11 novel miRNAs have proper precursor secondary structure and MFEI values of > 0.70. The secondary structure of the precursors was predicted using the RNAfold web server.17,27,28,36-42 The MFEI values were calculated utilizing the following formula:

The targets for conserved and novel miRNA were predicted using the psRNATarget tool.43,44 The steps involved in this study are shown in Figure 2.

Steps involved in analyses of P. sativum next-generation sequencing data.
Results
Analysis of small RNA transcriptome of P. sativum
As limited EST sequence information was present in the National Center for Biotechnology Information Expressed Sequence Tags database, it is highly impossible to identify P. sativum miRNAs merely based on computational analysis. Next-generation high-throughput sequencing is a robust tool for the de novo identification of miRNA present in P. sativum.
By utilizing high-throughput Illumina sequencing in this study, a total of 12,713,919 raw reads were generated from the small RNA library of garden pea leaves tissue. After filtering the low-quality reads, adapters, and redundant sequences along with rRNA (2,106), tRNA (424), mRNA (75,037), snRNA (1,024), and snoRNA (999), a total of 1,184,724 unique reads with lengths ranging from 16 to 40 nucleotides were retained for further analysis. It was observed that in P. sativum young leaves tissue small RNA library, the size of the small RNA was unevenly distributed as shown in Figure 3.

Small RNA length and reads distribution detected in P. sativum young leaves tissue using Illumina Next Generation Sequencing.
Identification of conserved miRNAs
To identify conserved miRNAs in P. sativum, Glycine max reference genome was downloaded from EnsemblPlants (https://plants.ensembl.org/). The unique sequences ⩾16 bp and ⩽40 bp length were considered for further analysis. Using Bowtie 1.3.1, the reads were aligned to the Glycine max reference genome. Then, the aligned reads were separated. The reads were checked for rRNA, tRNA, snRNA, and snoRNA contamination using the Rfam database. Finally, the filtered reads were used for conserved miRNA prediction. Using the CD-HIT program, the reads were made unique and the read count profile was generated.
Using the NCBI-blast-2.13.0 + program, a homology search of the unique miRNA reads was performed against matured miRNA sequences retrieved from miRbase-22. A total of 254 miRNA belonging to 56 different families were identified. miRNA family analysis revealed MIR-156 family was the most abundant, followed by MIR-166, MIR-159, etc. All the identified conserved miRNAs in P. sativum were shown in Table 1.
Conserved miRNAs identified in P. sativum.

The rest of the information related to the conserved miRNAs such as length, reference miRNA, read count, percent identity, alignment length, mismatches, and E value were attached as supplementary data S1.
Identification of novel miRNA
After the identification of conserved miRNA, the unaligned reads were used to predict novel miRNA using MIREAP software. The software integrates small RNA depth and position with the miRNA biogenesis model for detecting miRNAs from NGS small RNA libraries. A total of 11 novel miRNAs were identified, and the read count of these miRNA candidates varies from 20 to 5. psa-m0029-3p miRNA has the highest number of read counts. The MFEI value of all the novel miRNA precursors ranges from 0.72 to 1.40 ruling out the possibility of other types of RNAs like mRNAs (0.62–0.66), tRNAs (0.64), and rRNAs (0.59). 45 The novel miRNAs detected in P. sativum young leaf sample were shown in Table 2. The red color sequences in the precursor sequence represent mature miRNA. The rest of the information related to the novel miRNAs such as strand, length, read count, and MFEI were attached as supplementary data S2.
Potential novel miRNA identified in P. sativum.

The secondary structure of the novel miRNA precursor sequences was predicted using RNAfold WebServer (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi).46,47 Figure 4 shows novel psa-m0023-3p miRNA and psa-m0028-5p generated from RNAfold WebServer.

Secondary structure of novel psa-m0023-3p miRNA and psa-m0028-5p miRNA.
Predicting target for conserved and novel miRNA of P. sativum
To understand the functions of the identified P. sativum miRNAs, potential targets were predicted using psRNATarget tool (https://www.zhaolab.org/psRNATarget/). miRNAs with copy number ⩾5 were considered for target prediction. Pea mRNA and EST sequences were downloaded from NCBI and used as targets in psRNATarget tool with default parameters.
In this study, a total of 12,838 corresponding potential target gene products of P. sativum miRNAs were detected (9,715 target gene products for conserved miRNA and 1,064 target gene products for novel miRNA). Approximately 83% of the conserved miRNA directly cleaves the target mRNA whereas the rest of the conserved miRNA performs translational repression on the target mRNA. As 11 novel miRNAs were identified, some of them directly cleave the target mRNA and some perform translational repression.
In the conserved miRNAs, psa-miR1527 (191) has the highest number of potential targets, followed by psa-miR5261 (171) and psa-miR396 g-3p (165), whereas in novel miRNAs psa-m0025-3p (161) has the higher number of targets, followed by psa-m0028-5p (145) and psa-m0016-3p (121).
Functional annotation of the miRNA target was performed using Blast2Go software. The GO (Gene Ontology) terms were obtained. The miRNA target gene products along with GO_ID were categorized into biological processes, cellular components, and molecular functions.48,49 All the data related to conserved and novel miRNA targets, miRNA target gene products, and gene ontology functional categorization were attached as
Discussion
Over the past few years, NGS or deep sequencing technologies have been used in different molecular biology studies.15,50 Various studies have been conducted to identify conserved and novel miRNAs in different plant species; however, miRNA and their target remain unknown in an economically important plant such as garden pea.
The small RNA size distribution pattern in P. sativum young leaves shows that 63% of small RNA reads lie between 20 and 24 nucleotides and 24 nucleotides length small RNA reads are dominant (41%) in the entire sRNA transcriptome. A large number of sequencing reads of miRNA belonging to MIR-156, MIR-159, and MIR-166 families support the deep sequencing data in other leguminous species such as Medicago and peanut.51,52
Conversely, in wheat and rice plant species, MIR-169 family sequencing reads are high in number and MIR-156 family reads were very low in abundance indicating the presence of species-specific expression profile of miRNA. 53 All the detected novel miRNAs had MFEI value > 72 and appropriate secondary structures.
The variation in sequencing reads numbers of different members or isoforms of a miRNA family in P. sativum indicates that the regulatory role of the isoform is dominant during the particular stage of development. For instance, the MIR-172 family is likely to play a significant role in phase transformation and floral organ development in various plant species, and the reads number of psa-miR172a (64) belonging to the MIR-172 family was high compared to other isoforms of the family. 54 The comparative study of various miRNA isoforms present in a miRNA family during a particular stage of development or at specific conditions will provide valuable insights into the role-played by the miRNA in growth and development.
The lack of funding was the major limitation of the study. As leaf tissue contains a large repertoire of miRNAs;55,56 therefore, I have focused primarily on garden pea leaves in this study. It would be interesting to study the distribution of the identified miRNA in roots, stems, and other important organs and tissues of garden pea plants under various stress conditions to understand the regulatory mechanism of miRNAs and to develop strategies that will help in enhancing the production of garden pea crops.
Conclusions
In conclusion, in this study, a small-RNA library was prepared by high-throughput sequencing of P. sativum leaves to identify the conserved and novel miRNAs along with their potential targets. This study provides the first report on the identification of conserved and novel miRNAs in P. sativum leaves by deep sequencing. The valuable information obtained from this study will provide a strong base for the researchers to select the candidate miRNA or MIR family in P. sativum plants and check the regulatory role-played by the miRNA in response to biotic and abiotic stress, growth, and development. In addition, this study will enlighten our understanding of miRNA-mediated post-transcriptional gene regulation and could be helpful in the annotation of the genome.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322231162777 – Supplemental material for Identification of Conserved and Novel MicroRNAs with their Targets in Garden Pea (Pisum Sativum L.) Leaves by High-Throughput Sequencing
Supplemental material, sj-docx-1-bbi-10.1177_11779322231162777 for Identification of Conserved and Novel MicroRNAs with their Targets in Garden Pea (Pisum Sativum L.) Leaves by High-Throughput Sequencing by Qurshid Hasan Khan in Bioinformatics and Biology Insights
Supplemental Material
sj-docx-2-bbi-10.1177_11779322231162777 – Supplemental material for Identification of Conserved and Novel MicroRNAs with their Targets in Garden Pea (Pisum Sativum L.) Leaves by High-Throughput Sequencing
Supplemental material, sj-docx-2-bbi-10.1177_11779322231162777 for Identification of Conserved and Novel MicroRNAs with their Targets in Garden Pea (Pisum Sativum L.) Leaves by High-Throughput Sequencing by Qurshid Hasan Khan in Bioinformatics and Biology Insights
Supplemental Material
sj-xlsx-3-bbi-10.1177_11779322231162777 – Supplemental material for Identification of Conserved and Novel MicroRNAs with their Targets in Garden Pea (Pisum Sativum L.) Leaves by High-Throughput Sequencing
Supplemental material, sj-xlsx-3-bbi-10.1177_11779322231162777 for Identification of Conserved and Novel MicroRNAs with their Targets in Garden Pea (Pisum Sativum L.) Leaves by High-Throughput Sequencing by Qurshid Hasan Khan in Bioinformatics and Biology Insights
Supplemental Material
sj-xlsx-4-bbi-10.1177_11779322231162777 – Supplemental material for Identification of Conserved and Novel MicroRNAs with their Targets in Garden Pea (Pisum Sativum L.) Leaves by High-Throughput Sequencing
Supplemental material, sj-xlsx-4-bbi-10.1177_11779322231162777 for Identification of Conserved and Novel MicroRNAs with their Targets in Garden Pea (Pisum Sativum L.) Leaves by High-Throughput Sequencing by Qurshid Hasan Khan in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
I am thankful to the Department of Plant Sciences and School of Life Science, University of Hyderabad, for their support and cooperation. I would like to express my deepest gratitude to Prof. Ch. Venkataramana, Department of Plant Sciences, University of Hyderabad, and Dr. Satendra Kumar Mangrauthia, Senior Scientist, IIRR, for their support.
Declaration Of Conflicting Interests:
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The author declares that he has no known competing financial interests or personal relationships that could have appeared to influence the work reported in this article.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by BIRAC SRISTI Appreciation Grant.
Author Contributions
Q.H.K. conceived, planned, and carried out the experiment, processed the experimental data, performed the analysis and interpretation of results, and prepared the manuscript.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.