
Abstract
Multiple myeloma (MM) is a hematological malignancy in which monoclonal plasma cells multiply in the bone marrow and monoclonal immunoglobulins are overproduced in older people. Several molecular and cytogenetic advances allow scientists to identify several genetic and chromosomal abnormalities that cause the disease. The comprehension of the pathophysiology of MM requires an understanding of the characteristics of malignant clones and the changes in the bone marrow microenvironment. This study aims to identify the central genes and to determine the key signaling pathways in MM by in silico approaches. A list of 114 differentially expressed genes (DEGs) is important in the prognosis of MM. The DEGs are collected from scientific publications and databases (https://www.ncbi.nlm.nih.gov/). These data are analyzed by Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) software (https://string-db.org/) through the construction of protein-protein interaction (PPI) networks and enrichment analysis of the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, by CytoHubba, AutoAnnotate, Bingo Apps plugins in Cytoscape software (https://cytoscape.org/) and by DAVID database (https://david.ncifcrf.gov/). The analysis of the results shows that there are 7 core genes, including TP53; MYC; CDND1; IL6; UBA52; EZH2, and MDM2. These top genes appear to play a role in the promotion and progression of MM. According to functional enrichment analysis, these genes are mainly involved in the following signaling pathways: Epstein-Barr virus infection, microRNA pathway, PI3K-Akt signaling pathway, and p53 signaling pathway. Several crucial genes, including TP53, MYC, CDND1, IL6, UBA52, EZH2, and MDM2, are significantly correlated with MM, which may exert their role in the onset and evolution of MM.
Keywords
Introduction
Multiple myeloma (MM) is one of the incurable hematological diseases, characterized by the proliferation of abnormal plasma cells with distinct cytogenetic characteristics in the bone marrow, representing about 10% to 15% of all hematopoietic tumors. 1 The International Agency for Research on Cancer (IARC) estimates a worldwide incidence of 160 000 cases and a worldwide mortality of 106 000 patients in 2018. 2 However, men are much more affected by this disease than women. Moreover, the median age of patients at diagnosis is about 66 to 70 years old, with 37% of patients being younger than 65 years old. 3
Multiple myeloma is a complex genomic landscape and it is characterized by many types of chromosomal aberrations. Hyperdiploidy and immunoglobulin heavy chain (IGH) translocations are included as early occurrences. They are present in the precursor stages of monoclonal gammopathy of undetermined significance (MGUS) and latent multiple myeloma and are completely clonal in the majority of cases. Hyperdiploidy is considered as the first type of copy number alteration commonly seen in MM. It is usually associated with a standard risk prognosis. It is also characterized by trisomies of 3 or more of the odd-numbered chromosomes, namely, 3, 5, 7, 9, 11, 15, 19, and 21, whereas the most common IGH translocations are t(4; 14), t(11; 14), t(14; 16), t(14; 20), and t(6; 14) and its prognostic impact depends largely on the partner chromosome. 4 Other alterations in copy number, gains, and losses of the whole chromosome or part of the chromosome occur in the later stages of the disease trajectory and are not considered triggering events. The exception is the gain of the first copy of 1q, which appears to be an early event, whereas subsequent additional gains of 1q are later events. Common gains and losses include del 1p, gain 1q, del 13/13q, del 17p, in addition to del 16q and del 12p. Moreover, point mutations occur later in MM and commonly affect the MAPK pathway, NFKB pathway, DNA damage and repair, plasma cell differentiation, MYC activation, regulation of gene expression, and the cell cycle pathway. 5 These mutations can arise in various subclones, and their influence on disease progression varies depending on environmental factors. 6 KRAS, NRAS, and BRAF were among the most commonly mutated genes involved in the MAPK pathway, where hot-spot activating mutations have been discovered and were present in roughly 40% to 50% of patients with newly diagnosed multiple myeloma. Recurrent mutations have also been observed in FAM46C, CYLD, DIS3, IRF4, TRAF3, TP53, RB1, LTB, SP140, and more, and mutations in therapeutic targets, namely, lenalidomide targets (CRBN, IKZF1, and IKZF3), proteasome subunits, and steroid receptor (NR3C1), can induce treatment resistance. The combination of all these chromosomal aberrations and gene mutations leads to a differential gene expression profile in the plasma cells of patients with MM. 7
Bioinformatics is one of the newest fields of biological research, which should be broadly considered as the use of mathematics, statistics, and informatics to process and analyze biological data.8,9 In addition, it is an important element in laboratories that generate, analyze, store, and interpret data from molecular genetic tests. 10 The present study aims to predict interactions between differentially expressed genes (DEGs) to identify central genes and determine key signaling pathways in MM to understand its genetic heterogeneity.
Materials and Methods
To accomplish this integrative analysis, a pipeline was used (Figure 1).

Pipeline chart of all study analysis steps. DEG indicates differentially expressed genes; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; PPI, protein-protein interaction.
A set of 114 DEGs in myeloma plasma cells involved in the progression of the disease were collected from different scientific publications11 -47 and databases (https://www.ncbi.nlm.nih.gov/). The following search terms were used: multiple myeloma, gene expression, heterogeneity, mutational profiles, and genetic predisposition.
We used the GenBank database, available at the following link (https://www.ncbi.nlm.nih.gov/gene) and GeneCards (https://www.genecards.org/) to match each gene to its ID and functional annotation (Table 1).
Differentially expressed genes (DEGs) in multiple myeloma selected from various databases and scientific publications.
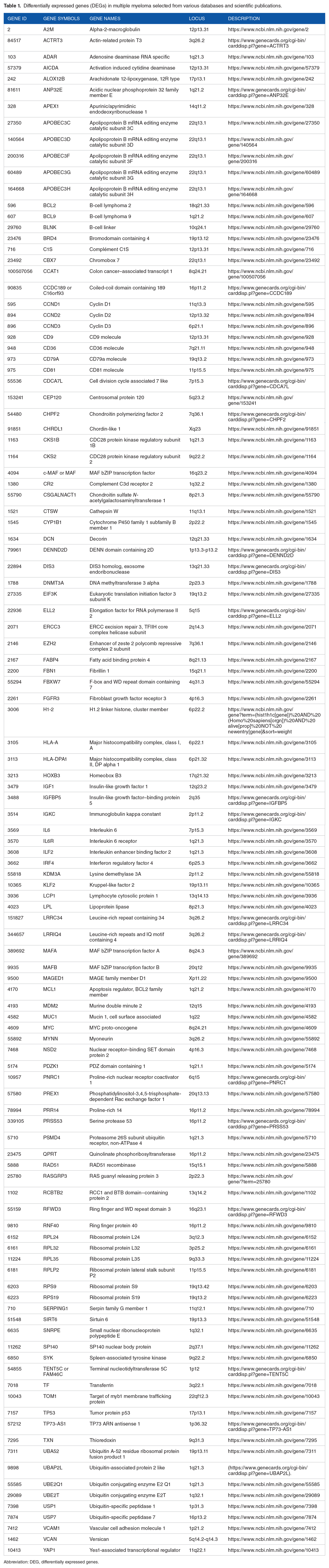
Abbreviation: DEG, differentially expressed genes.
Analysis Software
STRING software analysis
Analysis by STRING version 11.5 (https://string-db.org/) allowed us to reveal key genes and co-expressed genes in MM, to identify enriched biological terms, GO terms, and finally to determine key signaling pathways of core genes. FDR value ⩽ 0.05 was considered as the cut-off criterion, with a confidence score of 0.400 set as the cut-off criterion.
Determination of key genes
The input file, composed of 114 DEGs collected, has been submitted for analysis by STRING. The parameters retained for the analysis were the type of organism (Homo sapiens), type of network (evidence network), and confidence score (<0.400). The analysis was performed without enrichment. The core genes were selected based on their number of interactions with other genes (⩾20 interactions) and their location in the PPI network (at the center).
GO analysis
The GO enrichment analysis includes biological process, cellular component, and molecular function. GO terms have been analyzed from files corresponding to the GO enrichment, then downloaded from the software in tabular separated values (TSV) format, which can be opened in Excel as simple tabular text output. The top GO analysis results were selected with their significant values of FDR.
Identification of co-expressed genes
To determine the co-expressed genes, a file corresponding to the string interaction was downloaded from the software in TSV format, which can be opened in Excel as a simple tabular text output. We allow classifying the co-expressed genes by their scores, which indicates the level of association of the expression data and can also be determined by the presence of a black line between the nodes in the PPI network.
Determination of key signaling pathways
They were obtained from the Kyoto Encyclopedia of Genes and Genomes (KEGG) process in String software and selected with their significant FDR value ⩽ 0.05.
Cytoscape software analysis
To verify the results obtained by String software, Cytoscape version 2.3.8 (https://cytoscape.org/) was used, considering the P value < .05 and a confidence score of 0.400 as cut-off criteria.
Key genes identification
The TSV file of the gene’s interaction was imported into Cytoscape software. The CytoHubba Cytoscape plugin was used to classify the network nodes according to their characteristics. The degree is one of the topological analysis methods provided by CytoHubba, allowing us to observe target genes with higher degrees, which could constitute key genes.
GO enrichment
The Cytoscape BiNGO plugin was used to provide the GO enrichment. The most significant GO terms were selected with their P value < .05 and their number of annotated genes.
Network clusters
Cytoscape software groups PPI networks generated by STRING into clusters, taking the P value < .05 and a confidence score of 0.400 as cut-off criteria. ClusterMaker Apps from AutoAnnotate application was used to perform Markov Clustering (MCL) of the protein network. Markov Clustering makes it possible to visualize the enriched terms in the form of circular plots on the nodes of the network.
DAVID functional annotation bioinformatics microarray analysis
The DAVID database version 6.8 (https://david.ncifcrf.gov/) was used to perform KEGG pathway enrichment, to determine the most significant signaling pathways and compare them with those obtained by STRING. The data were pasted as a list of official gene symbols into the DAVID database.
Results
Network analysis
The network analysis of the set of 114 DEG revealed that 110 were annotated by STRING (Figure 2). A central network was obtained encompassing 99 genes, and a small network at the periphery, grouping together 2 genes, APOBEC3H and APOBEC3C. In addition, 9 genes have been outside the core network: RASGRP3; CEP120; MAGED1; CHRDL1; PNRC1; ALOX12B; PREX1; RCBTB2; DENND2D, and 7 core genes constitute the network engine: TP53; MYC CDND1; IL6; UBA52; EZH2; MDM2. In this functional network, all 101 genes represented various interactions, which may be explained by functional links between them.

Network of protein-protein interaction. The network view (evidence view) summarizes the set of predicted associations for a group of 110 genes. The nodes of the network are the gene product, and the edges represent the predicted functional associations. The edges are represented by lines of different colors that indicate the type of interaction to predict the associations. Clicking on a node will give detailed information about the protein and clicking on an edge will display a detailed breakdown of the evidence.
Co-expression results
From the first network, we determined the co-expression related to the core target genes of MM (Table 2). Key genes have been identified by their scores, which indicate the level of association of the gene expression data. The nodes represented genes and the black line shown between the nodes in the PPI network indicated co-expression (Figure 2). A co-expression score existing between 2 genes represented it. The score indicated the level of association of expression data during a process. If 2 genes showed similar expression under different conditions, it was likely that they were jointly involved in the same process (one requiring the other).
Co-expressed between core genes.

The genes marked in bold signify co-expression between 2 key genes.
Among the identified core genes, some of them represented co-expression with each other, including CCND1 with MYC, EZH2 with MYC, EZH2 with TP53, and MYC with TP53. In addition, they also represented co-expression with other genes in the functional network, which were presented in Table 1.
GO enrichment
GO terms were classified in the STRING software according to the value of the false discovery rate and the number of core genes they contained. Moreover, they were classified according to the number of annotated genes in each GO term. Thus, the most important ones had a high number of key genes involved in the development of MM. The GO analysis allowed the obtaining of a total of 448 GO items, including 392 biological process (BP) entries, 23 molecular function (MF) entries, and 33 cellular component (CC) entries (Figure 3).

Gene Ontology enrichment. The diagram represents the most significant GO terms according to the number of genes involved in the network, which are indicated in parenthesis in the diagram.
The most significant biological process determined with FDR values of 7.55e-09; 1.26e-08; 1.26e-08 were as follows: Cellular macromolecule metabolic process (GO:0044260) (64/114 genes), regulation of gene (70/114 genes), respectively.
The most significant molecular function observed with FDR values of 0.00011; 0.00019; 0.00019; were as follows: RNA binding (GO:0003723) (28/114 genes), nucleic acid binding (GO:0003676) (46/114 genes), and protein binding (GO:0005515) (66/114 genes), respectively.
Potentially important target genes have been expressed in the membrane-bounded organelle (GO:0043227), organelle (GO:0043226), and intracellular organelle (GO:0043229), which have a number of annotated genes of 97/114; 100/114; 96/114 and FDR values of 2.80e-06; 1.45e-05; and 1.45e-05, respectively.
Identification of KEGG pathways
KEGG pathway enrichment analyses were performed to reveal potential signaling pathways in the 114 DEGs (Table 3). They were significantly enriched in Epstein-Barr virus (EBV) infection (hsa05169), MicroRNAs in cancer (hsa05206), PI3K-Akt signaling pathway (hsa04151), and p53 signaling pathway (hsa04115), which were considered as the major pathways involved during the development of MM.
KEGG pathways enrichment.

Abbreviation: KEGG, Kyoto Encyclopedia of Genes and Genomes.
The table represents the most important KEGG pathways, selected with their significant FDR value ⩽ 0.05 and their number of genes annotated in the functional network.
Cytoscape software analysis
Identification of key genes
Cytoscape analysis showed 110 annotated genes. CytoHubba apps from the Cytoscape program allowed users to determine the top genes with the “degree” method, where the target genes have the highest degrees, and which are often key target genes (Figure 4). Each node showed a different color depth depending on its own degree (the darker the color, the higher the degree). The network provided 7 important key target genes involved in the regulation of many other genes in the PPI network, which may have potential therapeutic targets in MM.

Network of genes interaction degree. Proteins with a higher degree of importance are more likely to be essential.
GO enrichment by BINGO plugin
The analysis by the BINGO apps in Cytoscape allowed identifying a set of GO terms with a high number of annotated genes and a significant P value (Supplementary Table S1). These results supported the results obtained by STRING software, and the most GO common were:—Biological process: GO ID
Network clusters
The 110 annotated genes were involved in 13 clusters, of which 12 were linked, forming a large network, and 1 remained outside this large network named Apolipoprotein deaminase single, with 2 genes (APOBEC3H and APOBEC3C) (Figure 5). The subgroups of this large network were named according to their protein annotations, and they were classified according to the number of genes involved: Susceptibility apoptosis cell is the largest subgroup and the most important, containing 51 genes, of which 6 were top genes (TP53, c-MYC, CCND1, EzH2, IL6, and MDM2);
Protein us4 ribosomal with 10 genes of which one (UBA52) is the top gene;
Reattaching heterochromatin mitotic with 8 genes;
Chondroitin proteoglycan microfibrils with 5 genes;
Proteinase c1s c1r with 4 genes;
na chloride scarb1 with 4 genes;
Somatomedin insulin higher with 3 genes;
Single deaminase independent with 3 genes;
Removes h2a h2afz with 3 genes;
Exosome 3’ untranslated with 3 genes;
dm mhc class with 2 genes;
Cytolytic release cytochrome with 2 genes.

Clustered protein association network. The cluster network provides 13 groups (clusters) of all 114 myeloma gene sets. Each node presents a gene or gene product, the color of the node indicates the 3-dimensional structure of the protein. Each gene cluster indicates a biological function.
DAVID database results
To validate the results obtained by STRING and Cytoscape, an analysis by the DAVID software was carried out. The results obtained showed 10 GO terms representing 10 signaling pathways classified according to their P value and the number of genes involved in them (Table 4). The most significant signaling pathways were EBV infection, PI3K-Akt signaling pathway, MicroRNAs in cancer, and the p53 signaling pathway. These results consolidated those obtained by STRING software.
KEGG pathways enrichment by DAVID.
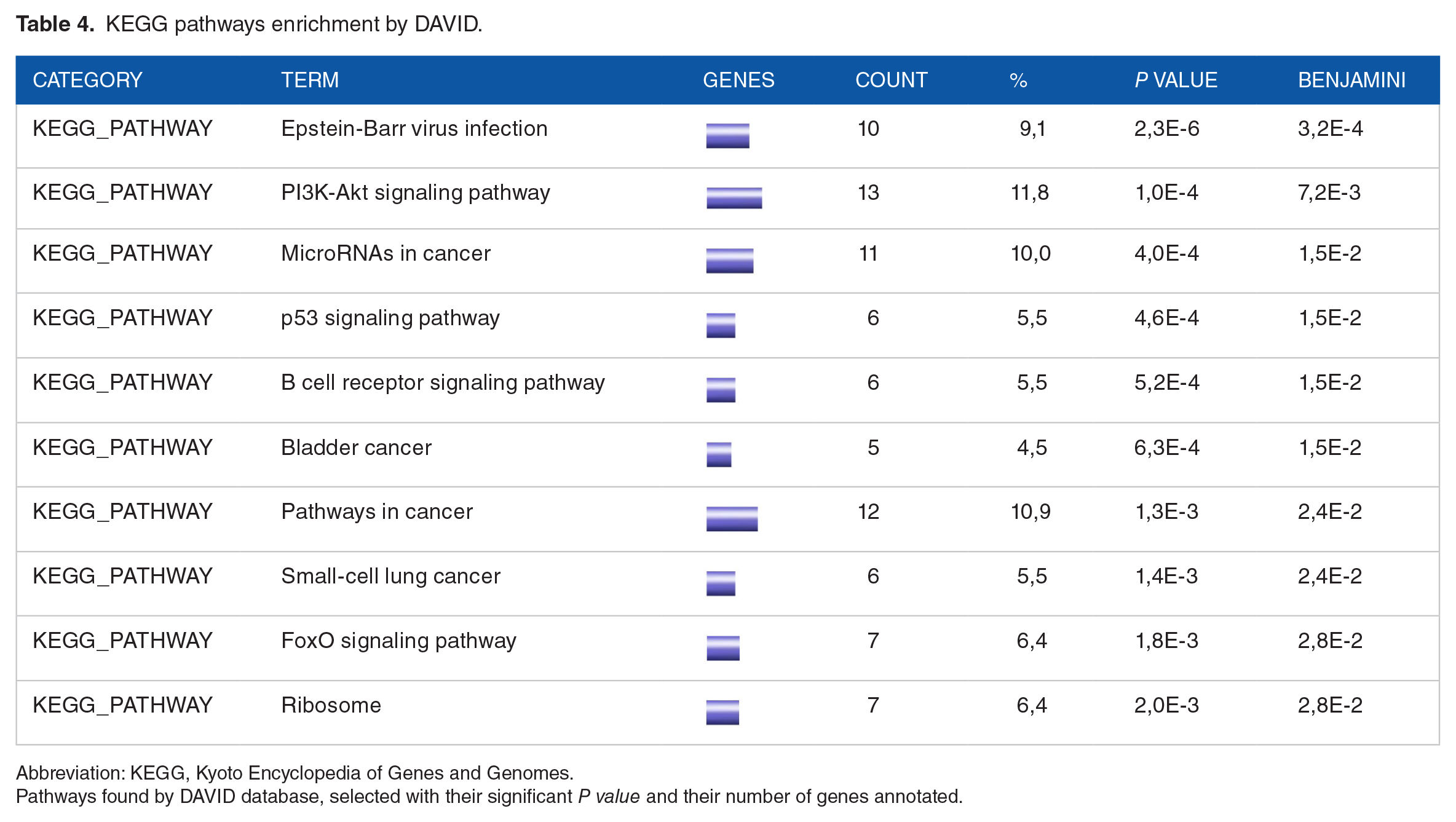
Abbreviation: KEGG, Kyoto Encyclopedia of Genes and Genomes.
Pathways found by DAVID database, selected with their significant P value and their number of genes annotated.
Discussion
In this study, we performed a bioinformatics analysis of a biological data set to identify the key genes and signaling pathways involved during the development of MM. The functional PPI network resulting from the STRING database and Cytoscape software identified that among 114 DEGs introduced, 7 are considered as key genes that represented a high connectivity in the network. Including TP53; MYC, CDND1, IL6, UBA52, EZH2, and MDM2. The CCND1 with MYC, EZH2 with MYC, EZH2 with TP53, and MYC with TP53 are co-expressed, which can be explained by their complementarity and involvement in the same or different processes during the transformation of normal plasma cells into myeloma plasma cells (MM evolution). 5
Indeed, the study of the co-expression of cell cycle genes has shown the presence of a dynamic balance between the co-expression of sets of genes activating and inhibiting the cell cycle (CDK/cyclin-dependent kinases and CDK1) in MM. 48 The clustering analysis showed that the susceptibility apoptosis cell cluster (the largest subgroup and the most important), containing 6 top genes (TP53, c-MYC, CCND1, EzH2, IL6, and MDM2), could be implicated in the development of MM.5,16,20,49,50 Nevertheless, the UBA52 gene was found in the protein us4 ribosomal cluster. The ribosomal us4 protein has multiple functions, including mRNA decoding, initiation of small ribosomal subunit aggregation by binding directly to the 16S rRNA and translation repression. It was also involved in the anti-termination activities of transcription. 51
The ontological analysis revealed the involvement of these 7 hub genes of the functional network in BP related to cellular macromolecular metabolic process, regulation of gene expression, macromolecular metabolic process, and positive regulation of the biological process. These hub genes were the major regulators of these 4 important processes that can be altered during the development of MM.16,49,52 -54 The CC results suggested that the 7 hub genes were mainly scattered throughout the cell, including the nucleus, mitochondria, plastids, vacuoles, vesicles, ribosomes, and the cytoskeleton. Then, the MF reflected the involvement of hub genes in RNA binding, nucleic acid binding, and protein binding. The alteration in the expression of one or more of these key genes may lead to deregulation of these processes, which can be essential for the cell life cycle and their deregulation may play a role in the development of MM.16,20,49,50
The
In the PPI network,
KEGG pathway enrichment analysis showed that the 7 key genes were significantly enriched in EBV infection (hsa05169), microRNAs in cancer (hsa05206), PI3K-Akt signaling pathway (hsa04151) and p53 signaling pathway (hsa04115). Many studies have been in concordance with our results, which demonstrated the existence of an association between EBV infection and MM development, in which a polymerase chain reaction was performed and revealed the presence of an elevated level of EBV DNA in patients with MM compared with healthy controls.75,76 MicroRNAs (miRNAs) are small noncoding RNAs aberrantly expressed in solid and hematopoietic malignancies, where they play a central role as post-transcriptional regulators of gene expression. 77 Recently, some studies have demonstrated the efficacy of miRNAs as specific and sensitive biomarkers for the classification, prognosis, and diagnosis of human cancer. 78 Jones et al 79 discovered 3 miRNAs in serum, miR-1308, miR-1246, and miR 720, which have the potential to be used as diagnostic biomarkers in myeloma. They demonstrated that the joint use of miR-1308 and miR-720 provided a powerful diagnostic tool for distinguishing normal healthy controls from patients with MGUS/myeloma. Furthermore, the combination of miR-1246 and miR-1308 can distinguish patients with MGUS from those with myeloma. 79 The PI3K/Akt pathway has attracted considerable attention as a promising therapeutic target in MM. 80 A study conducted on the PI3K/Akt/mTOR pathway (mammalian target of rapamycin) by Vijay Ramakrishnan and Shaji Kumar has shown that it played a critical role in the biology of diseases. This pathway was aberrantly activated in a large proportion of patients with MM by multiple mechanisms. It may also play a role in resistance to several existing therapies, making it a central pathway in the pathophysiology of MM. 81 The study by Vikova et al 82 showed after whole-exome sequencing on a large cohort of 30 human MM cell lines, representative of a large molecular heterogeneity of MM, and 8 control samples, that many canonical pathways known to be implicated in the proliferation and survival of MM cells have been mutated, including PI3K-AKT. In addition, 76% of myeloma cell lines had mutations in the p53 cell cycle pathway genes. 82
Conclusion
STRING, Cytoscape, and DAVID Software analyzed a set of 114 MM DEGs. TP53, MYC, CCND1, IL6, UBA52, EZH2, and MDM2 were identified as hub genes, which may play a key role in the pathogenesis of MM. They were implicated during different stages of disease progression. The 7 hub genes were identified as being significantly enriched in various pathways, particularly in EBV infection, microRNAs in cancer, the PI3K-Akt signaling pathway, and the p53 signaling pathway, which can be critical for the progression of myeloma. This predictive study can be a relevant tool for a better understanding of the evolution of MM in its complex microenvironment. Understanding the pathogenesis of MM could reveal new biomarkers of myeloma cells and promising potential therapeutic targets.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322221115545 – Supplemental material for Multiple Myeloma: Bioinformatic Analysis for Identification of Key Genes and Pathways
Supplemental material, sj-docx-1-bbi-10.1177_11779322221115545 for Multiple Myeloma: Bioinformatic Analysis for Identification of Key Genes and Pathways by Chaimaa Saadoune, Badreddine Nouadi, Hasna Hamdaoui, Fatima Chegdani and Faiza Bennis in Bioinformatics and Biology Insights
Footnotes
Author Contributions
CS did the conceptualization, methodology, data collection, analysis and interpretation, wrote original draft. BN collected data, revised, and edited manuscript. HH revised and edited manuscript. FC did the conceptualization, wrote—revised and edited, supervision. FB did the conceptualization, wrote—revised and edited, supervision. All authors contributed to manuscript revision, read and approved the submitted version.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.