
Abstract
Differential expressions of certain genes during tumorigenesis may serve to identify novel manageable targets in the clinic. In this work with an integrated bioinformatics approach, we analyzed public microarray datasets from Gene Expression Omnibus (GEO) to explore the key differentially expressed genes (DEGs) in non-small cell lung cancer (NSCLC). We identified a total of 984 common DEGs in 252 healthy and 254 NSCLC gene expression samples. The top 10 DEGs as a result of pathway enrichment and protein–protein interaction analysis were further investigated for their prognostic performances. Among these, we identified high expressions of CDC20, AURKA, CDK1, EZH2, and CDKN2A genes that were associated with significantly poorer overall survival in NSCLC patients. On the contrary, high mRNA expressions of CBL, FYN, LRKK2, and SOCS2 were associated with a significantly better prognosis. Furthermore, our drug target analysis for these hub genes suggests a potential use of Trichostatin A, Pracinostat, TGX-221, PHA-793887, AG-879, and IMD0354 antineoplastic agents to reverse the expression of these DEGs in NSCLC patients.
Keywords
Introduction
Lung cancer is one of the deadliest diseases all around the world. The GLOBOCAN estimations by the International Agency for Research on Cancer predict approximately 13 million new cancer cases by the year 2040. Currently, lung cancer is the most commonly diagnosed form of cancer (11.6% of total cases) and the leading cause of cancer deaths (18.4% of total deaths). 1 Histologically, lung cancer can be divided into non-small cell lung cancer (NSCLC), which accounts for approximately 85% of lung cancer cases and small cell lung cancer. 2 The lack of early-stage symptoms and effective diagnostic markers restrain the treatment success in NSCLC. Thus, most of the patients are diagnosed at an advanced stage, and half of them have distant metastatic disease at initial diagnosis. 3 Over the last decade, there have been considerable improvements in chemotherapy, radiation therapy, surgery, and targeted therapy for lung cancer. Especially with the recent advances in molecular biology, significant progress has been made through molecule-targeted therapy in NSCLC. For instance, it has been shown that about 20% of Caucasian and 50% of Asian NSCLC patients had mutations on their EGFR (epidermal growth factor receptor) genes. 4 However, with the application of EGFR targeting small tyrosine kinase inhibitors (EGFR-TKIs) erlotinib and gefitinib, both response rate and median survival of these patients were found to be improved. 5 Also, approximately 7% of NSCLC patients bear an activated ALK gene, but treatment with ALK inhibitor crizotinib has been shown to improve both response and 6-month progression-free survival rates in these patients. 6 Nonetheless, the 5-year survival rates of NSCLC patients remain low with a poor prognosis due to the development of intrinsic or acquired chemoresistance against therapeutic drugs. 7
Non-small cell lung cancer is a result of the accumulation of several genetic and epigenetic modifications, which could have originated from multiple reasons. 8 The discovery and characterization of new prognostic or diagnostic markers together with enhanced therapeutic approaches for NSCLC are of top priority for the successful treatment of this disease. Unfortunately, information on the heterogeneous nature of the tumor and the involvement of affecting factors in the process of NSCLC tumor development are far from completely resolved. Therefore, it is highly important to shed light on the molecular mechanisms governing the pathogenesis of NSCLC and to identify effective diagnostic and/or prognostic biomarkers for novel treatment options. High-throughput technologies such as microarrays and integrated bioinformatics methods are used to obtain gene alterations during tumorigenesis and to identify novel prognostic markers in patients with cancer.9,10 For instance, in a recent study, Huang and Gao 11 have demonstrated that CDC20, CENPF, KIP2C, and ZWINT genes were differentially expressed in NSCLC tissues. Similarly, Xiao et al 12 have identified CCNB1, CCNA2, CEP55, PBK, and HMMR as hub genes and key differentially expressed genes (DEGs) associated with NSCLC by bioinformatics analyses. Interestingly, Wang et al 13 have shown CCND1 as the most enriched gene and a potential prognostic biomarker in NSCLC through a gene set enrichment analysis. More recently, Zhang et al 14 have identified TOP2A, CCNB1, BIRC5, and TTK as well as miR-21-5p and miR-31-5p to be significantly associated with NSCLC prognosis through an integrative analysis of mRNA and miRNA expression profiles. Nevertheless, the genes that are discovered by 1 cohort might be difficult to be identified in other cohorts. 15 For this reason, it is essential to validate genes in several independent studies.
In this study, we sought to identify potential therapeutic targets or prognostic biomarkers among the DEGs associated with NSCLC through an integrated bioinformatics approach. For this purpose, we retrieved 4 different microarray datasets from Gene Expression Omnibus (GEO) database and screened for DEGs between NSCLC tumor and neighboring normal tissues. After gene set enrichment analysis to identify associated biological processes, a protein–protein interaction (PPI) network analysis was performed to elucidate potential key DEGs. We also explored the significance of candidate key DEGs and their correlation to patient prognosis through survival analysis. Finally, potential therapeutic drugs that may target and reverse the expression of these key DEGs were predicted by using the L1000CDS2 signature search engine.
Materials and Methods
Microarray datasets
A comprehensive database search was conducted for identifying appropriate datasets including NSCLC tumor tissue and matched adjacent normal samples from the public GEO database. 16 To avoid microarray platform differences, the datasets originating from Affymetrix microarrays utilizing Human Genome U133 Plus 2.0 chips (Thermo Fisher Scientific, Inc., Waltham, MA, USA) were selected. Four datasets with accession numbers GSE18842, GSE19804, GSE27262, and GSE102287 were identified and downloaded for further analysis (Table 1). GSE19804 18 included 60 pairs of NSCLC tumor and matched adjacent normal lung tissue, while GSE18842 17 comprised 46 NSCLC tumors and 45 paired controls, GSE27262 19 contained 25 tumors and normal tissue pairs from stage I lung adenocarcinoma, and GSE102287 20 contained 66 matched NSCLC tumor and normal tissues. A total of 506 gene expression samples including 252 healthy and 254 NSCLC tissues were evaluated in this study.
Transcriptome datasets employed in the present study.

Abbreviation: GEO, Gene Expression Omnibus.
Data preprocessing and identification of DEGs
To identify DEGs, raw data in the form of CEL files were downloaded from the GEO database. Affy package of R/Bioconductor platform (version 3.6) was utilized, and the gene expression datasets were normalized by using the Robust Multi-Array (RMA) techniques. 21 Linear models for microarray data (LIMMA) method 22 was used in statistical analysis of each dataset by comparing gene expressions in NSCLC tissues with neighboring healthy tissues to identify DEGs. Differentially expressed genes in each dataset were selected according to computed P values <.05. The regulatory pattern of each DEG was determined by fold changes (FCs) of up- and downregulation (FC >2.0 and <0.5, respectively), and at least 50% change was considered significant.
Functional and pathway enrichment analysis
To identify functional annotations significantly associated with the gene products, pathway enrichment analyses were performed using ConsensusPathDB. 23 Genomic, chemical, and systematic functional information of DEG pathways were provided by Kyoto Encyclopedia of Genes and Genomes (KEGG). 24 For each of the predefined sets, a P value is calculated according to the hypergeometric test, and enrichment results with P < .01 were considered statistically significant.
PPI analysis
Human protein interactome containing 43 219 physical interactions between 2294 human proteins was collected from the BioGRID database. 25 The physical interactions of the proteins encoded by the common genes of all datasets were analyzed in Cytoscape 26 by the reconstruction of PPI networks. Hub genes were identified according to degree and betweenness scores.
Prognostic performance analysis
Survival analyses were performed by Kaplan-Meier Plotter web tool 27 using mRNA data to identify the prognostic performances of each PPI hub gene. The subjects were partitioned into low- and high-risk groups according to their hazard ratio index. The prognostic capabilities of the genes were characterized through Kaplan-Meier plots, and the log-rank P value <.05 was considered the cutoff value to describe statistical significance. In addition, prognostic performances of the genes were validated using datasets with available clinical information obtained from CAARRAY, GSE14814, GSE19188, GSE29013, GSE30219, GSE31210, GSE3141, GSE31908, GSE37745, GSE43580, GSE4573, GSE50081, GSE8894, and TCGA in total of 1926 samples.
Drug target potential analysis
L1000CDS2 28 is a signature search engine which lists small molecules that are predicted to mimic or reverse the input gene expression profiles utilizing LINCS L1000 data. 29 We performed L1000CDS2 analysis using the hub gene list as an input, to assess the potential of agents that may reverse the expression of up- or downregulated hub genes in NSCLC. Resultant small molecules were selected according to 2 main criteria: (1) perturbed human cells as lung cancer cell lines and (2) top 2 overlap scores.
Results
Transcriptomic signatures of NSCLC
We comparatively analyzed 4 distinct transcriptome datasets composed of NSCLC and healthy tissue samples. After preprocessing of all 4 datasets, DEGs were identified between tumor samples and matched normal tissues according to our threshold criteria (FC > 2.0 and FC < 0.5 with computed P < .05). We obtained 3381, 1581, 2488, and 1934 respective DEGs in GSE18842, GSE19804, GSE27262, and GSE102287 datasets. Among these, 1451, 455, 966, and 621 genes were upregulated, while 1951, 1075, 1537, and 1322 genes were downregulated in GSE18842, GSE19804, GSE27262, and GSE102287 datasets, respectively (Figure 1A and Supplementary Table 1). Combination and comparison of all 4 total DEG groups resulted in the identification of 984 common DEGs (Figure 1B), with 246 upregulated (Figure 1C) and 734 downregulated (Figure 1D) genes. It was determined that the number of downregulated genes was higher than the number of upregulated genes in all datasets.

DEGs in non-small cell lung cancer. (A) The distribution of DEGs in non-small cell lung cancer–associated transcriptome datasets. Downregulation and upregulation of DEGs were represented by blue and orange colors, respectively. (B) The Venn diagram represents the comparison of DEGs among all datasets. (C) The Venn diagram represents the comparison of upregulated DEGs among all datasets. (D) The Venn diagram represents the comparison of downregulated DEGs among all datasets. DEGs indicate differentially expressed genes.
Biological insights of common transcriptomic signatures
The pathway enrichment analysis based on situated common up- and downregulated DEGs indicated significant results for general cancer signaling, and metabolic and immune systems–related pathways. Our results showed that commonly upregulated genes were significantly enriched in 10 KEGG pathways, which included cell cycle, p53 signaling pathway, and extracellular matrix (ECM)–receptor interaction (Figure 2A). On the contrary, the downregulated common DEGs were enriched in 32 terms, including malaria, cell adhesion molecule, and cAMP signaling pathway (Figure 2B). The top 15 downregulated pathways were used in Figure 2B. The rest of the enriched terms can be provided upon request.

Statistically significant biological pathways of common DEGs: (A) upregulated pathways and (B) downregulated pathways. Up- and downregulated common DEGs list was used to obtain up- and downregulated pathways. In both cases, top 15 enriched pathways were represented. DEGs indicate differentially expressed genes.
NSCLC-specific PPI network
To identify hub genes and to reconstruct a PPI network, we utilized the BioGRID database and analyzed proteins from 984 identified DEGs with Cytoscape. In topological analyses of the NSCLC-specific PPI network, degree (local-based) and betweenness centrality (global-based) metrics 30 were used to identify the highly connected proteins, that is, hub proteins (Figure 3A), which might play an important role in cancer pathogenesis. According to their degree and betweenness scores, the top 10 hub genes with strong interactions with others were determined (Figure 3B). We selected Cbl Proto-oncogene (CBL), Enhancer of Zeste 2 Polycomb Repressive Complex 2 Subunit (EZH2), Cyclin Dependent Kinase 1 (CDK1), Cell Division Cycle 20 (CDC20), Cyclin Dependent Kinase Inhibitor 2A (CDKN2A), Aurora Kinase A (AURKA), FYN Proto-oncogene (FYN), ETS Transcription Factor ERG (ERG), Suppressor of Cytokine Signaling 2 (SOCS2), and Leucine Rich Repeat Kinase 2 (LRKK2) as the most significant nodes for further evaluation.

Non-small cell lung cancer-specific protein–protein interaction network and hub proteins. AURKA indicates Aurora Kinase A; CBL, Cbl Proto-oncogene; CDC20, Cell Division Cycle 20; CDK1, Cyclin Dependent Kinase 1; CDKN2A, Cyclin Dependent Kinase Inhibitor 2A; ERG, ETS Transcription Factor ERG; EZH2, Enhancer of Zeste 2 Polycomb Repressive Complex 2 Subunit; FYN, FYN Proto-oncogene; LRKK2, Leucine Rich Repeat Kinase 2; SOCS2, Suppressor of Cytokine Signaling 2.
Prognostic hub genes in NSCLC
To evaluate the prognostic performances of the top 10 hub genes, we performed a survival analysis by Kaplan-Meier Plotter. The overall survival times for patients with NSCLC were obtained according to the low and high expressions of each hub gene (Figure 4). Our results showed that high mRNA expression of CDC20 (Hazard ratio [HR], 1.82; confidence interval [CI], 1.6-2.07) as well as AURKA (HR, 1.52; CI, 1.33-1.72), CDK1 (HR, 1.4; CI, 1.23-1.59), EZH2 (HR, 1.31; CI, 1.15-1.48), and CDKN2A (HR, 1.29; CI, 1.13-1.46) were associated with significantly poorer overall survival (P < .05) for NSCLC patients. According to our statistical analysis, these genes were among the commonly upregulated genes with significantly higher expression FC levels in NSCLC samples in all datasets (Figure 5). On the contrary, high mRNA expression of CBL (HR, 0.73; CI, 0.62-0.86) as well as FYN (HR, 0.71; CI, 0.62-0.80), LRKK2 (HR, 0.62; CI, 0.52-0.73), and SOCS2 (HR, 0.62; CI, 0.53-0.74) were associated with significantly better prognosis (P < .05). Accordingly, expression levels of these genes were found to have significantly lower FC values in NSCLC tumor samples in all datasets (Figure 5). Therefore, it is logical to predict a poor prognosis associated with these 4 downregulated hub genes. Unfortunately, ERG gene was found to have no statistically significant prognostic potential in a total of 1926 NSCLC samples.
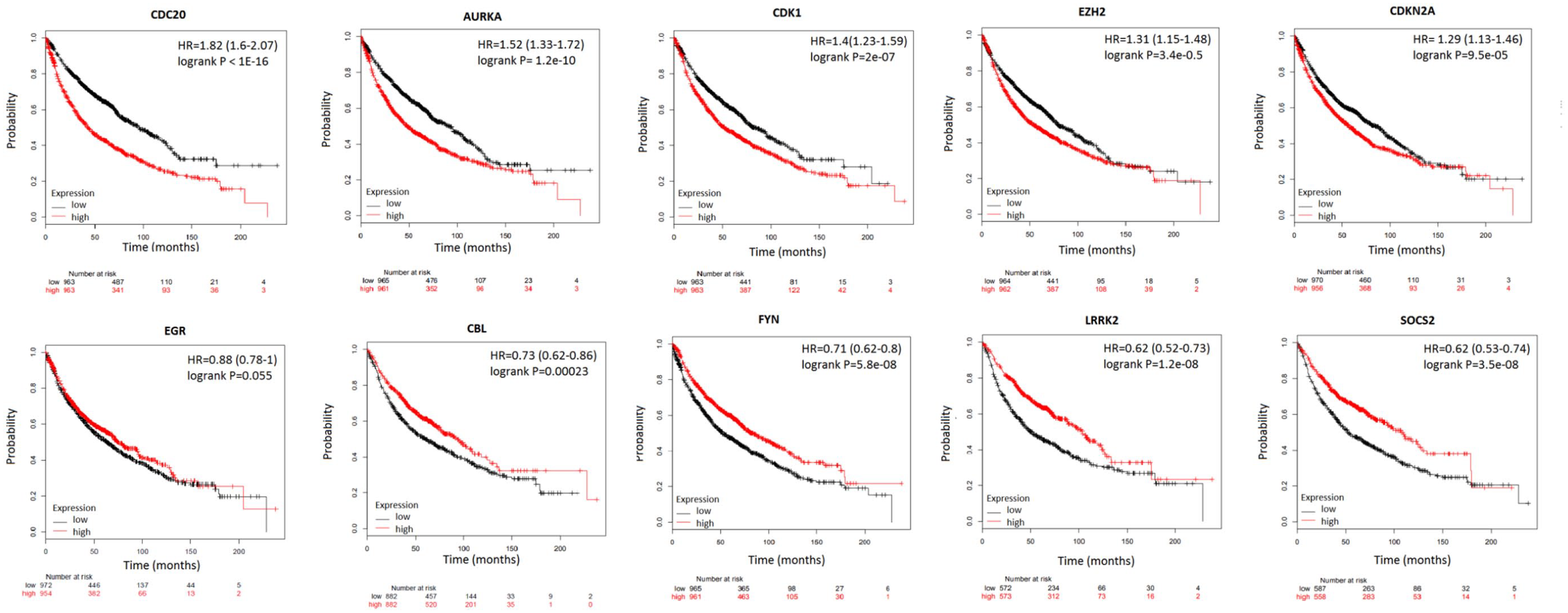
Survival analysis of hub differentially expressed genes. Kaplan-Meier Plot for patients with non-small cell lung cancer obtained from several independent databases. Downregulated expression is represented in a black plus sign, whereas upregulated expression in a red plus sign. AURKA indicates Aurora Kinase A; CBL, Cbl Proto-oncogene; CDC20, Cell Division Cycle 20; CDK1, Cyclin Dependent Kinase 1; ERG, ETS Transcription Factor ERG; FYN, FYN Proto-oncogene.

Fold changes of hub genes in each dataset. Red and blue grades represent upregulated and downregulated genes, respectively. AURKA indicates Aurora Kinase A; CBL, Cbl Proto-oncogene; CDC20, Cell Division Cycle 20; CDK1, Cyclin Dependent Kinase 1; ERG, ETS Transcription Factor ERG; EZH2, Enhancer of Zeste 2 Polycomb Repressive Complex 2 Subunit; FYN, FYN Proto-oncogene; SOCS2, Suppressor of Cytokine Signaling 2.
Drug target potential of the hub genes
To assess the potential drugs to therapeutically target identified hub proteins, we have utilized the LINCS L1000 connectivity map data and characteristic direction signature search engine L1000CDS2. The list of hub genes was entered into a web tool to search for substances that can reverse the expression changes in identified DEGs. Thirteen drugs or small molecules were identified, showing potential with an overlap score of >0.4 to reverse expression profiles on upregulated and/or downregulated gene expressions in lung cancer cell lines (Table 2). A considerable portion (6 out of 13) of the drugs was antineoplastic agents, which were suggested for the treatment and management of the cancers. The remaining drug candidates were originally used for purposes other than cancer treatments; however, they possess the potential to reverse the expression of the top 10 DEGs as shown in our analysis.
Potential drugs or small molecules that can reverse the expression change in identified hub proteins through LINCS L1000 connectivity map data.

Abbreviations: AURKA, Aurora Kinase A; CDC20, Cell Division Cycle 20; CDK1, Cyclin Dependent Kinase 1; CDKN2A, Cyclin Dependent Kinase Inhibitor 2A; DEGs, differentially expressed genes; EZH2, Enhancer of Zeste 2 Polycomb Repressive Complex 2 Subunit; FYN, FYN Proto-oncogene; SOCS2, Suppressor of Cytokine Signaling 2.
Discussion
Lung cancer tumorigenesis, progression, and metastasis are very complicated processes involving defects in multiple genes and cellular pathways. 31 Multiple interacting DEGs affecting other genes may constitute the core functional network of genes in promoting carcinogenesis. 32 For an improved diagnosis and treatment, it is crucial to discover these abnormal genes and understand their roles in the molecular mechanisms of NSCLC. Developments in microarray and high throughput technologies allow us to detect cancer etiology by examining abnormalities at the whole-genome level. These technologies have been widely used to predict the potential therapeutic targets for cancers. On the contrary, a combination of large data on chemical perturbational profiles of human cell lines with differential gene expression analysis may provide not only information on abnormal gene activities but also some important clues on therapeutic options to overcome these defects in NSCLC.
In this study, we identified 246 upregulated, 734 downregulated, and a total of 984 common DEGs between NSCLC and normal tissue samples by using public gene expression profiles of GSE18842, GSE19804, GSE27262, and GSE102287 datasets.
The original data on these datasets were generated to identify SEMA5A, 18 protein arginine methyltransferase 5, 33 and methylosome protein 50 19 as potential biomarkers in NSCLC. On the contrary, GSE18842 was generated to analyze DEGs as a function of tumor type, stage, and differentiation grade in NSLC. Keratin 15 and plakophilin 1 were identified as potentially good markers to distinguish squamous cell carcinoma, whereas a significant downregulation of desmoglein 3 was observed in early-stage tumor samples. 17 A comparative transcriptome profiling was utilized to show coding and noncoding RNA differences between NSCLC from different human races. In this study, the dataset GSE102287 was generated to show 40 different novel population-specific gene expressions such as ARL17A, LRCC37A3, and KANSL1. 20 Although the data were supported and validated by several wet-lab experimentations, none of these original works utilized a similar integrated bioinformatics approach. All 4 datasets re-analyzed in a single set for the first time in our study.
In our PPI network analysis, we identified a series of hub genes, including CDC20, AURKA, CDK1, EZH2, CDKN2A, CBL, FYN, LRKK2, SOCS2, and ERG as the top 10 significant nodes. Among them, CDC20 appears to act as a regulatory protein interacting with several other proteins such as mitotic spindle checkpoint protein MAD2L1 and anaphase-promoting complex/cyclosome (APC/C) at multiple points in the cell cycle. It is required for 2 microtubule-dependent processes, nuclear movement before anaphase and chromosome separation. 34 Disorders involving the CDC20 gene are mosaic variegated aneuploidy syndrome 1, neuronal ceroid lipofuscinosis, and prostate cancer. 35 CDC20 was identified as frequently upregulated in many types of malignancies including NSCLC and suppressed by p53 expression. 36 Importantly, CDC20 was identified among the key genes with prognostic value in NSCLC in several other studies using bioinformatics methods and similar datasets.37,38 Therefore, it may represent a potential molecular target and a critical molecular marker for NSCLC progression. All drugs on our candidate list were found to potentially interfere with CDC20 expression.
The second hub gene AURKA is another cell cycle–related protein which is a serine/threonine kinase that takes part in the regulation of cell cycle progression. During mitosis, it associates with centrosomes and spindle microtubules, and plays an essential role in various mitotic events such as the establishment of mitotic spindle, centrosome duplication and separation in addition to chromosomal alignment, spindle assembly checkpoint, and cytokinesis. In the checkpoint response pathways that are critical for oncogenic transformation of cells, it acts as a key regulatory component of p53/TP53 pathway by phosphorylating and stabilizing p53 itself. Interestingly, AURKA is required for the initial activation of CDK1 at centrosomes. 39 Aberrant expression of AURKA is associated with several cancer types, including colorectal cancer, laryngeal squamous cell carcinoma, atypical teratoid rhabdoid tumors, as well as tetraploidy syndrome. 40 In a recent study using gene expression meta-analysis integrated with neural network algorithms, AURKA was identified as the most obvious class of hub genes associated with NSCLC. 41 Furthermore, the amplification or activation of AURKA-induced impairment of the LKB1/AMPK axis was found to contribute to NSCLC initiation and progression, and suggests AURKA as a potential therapeutic target. 42 In a few bioinformatics studies using very similar integrative approach and some of the datasets used in this study, AURKA was suggested as one of the hub genes in NSCLC.43,44 CDK1 plays a key role in the control of the eukaryotic cell cycle by modulating the centrosome cycle as well as the mitotic onset. Besides the promotion of G2-M and G1S transitions, CDK1 regulates G1 progress via association with multiple interphase cyclins and it is required for entry into both S phase and mitosis in higher cells. In addition, as a master modulator in cell cycle onset and progression, it phosphorylates and activates numerous proteins, including CDC20 and EZH2. Loss of CDK1 activity or the aberrant expression of CDK1 is involved in G2 phase arrest in many tumor types. Therefore, an interest has been developed to search for potent CDK1 inhibitors and it comprises an attractive target in oncology. 45 Our results together with some recent other bioinformatics studies utilizing GSE18842 dataset 46 support the critical role of CDK1 as an important oncological target in NSCLC as well. Among the other identified hub genes, EZH2 is a polycomb group protein and the core catalytic subunit of a protein complex that participates in transcriptional repression of the affected target genes by methylated lysines on histone H3 through multiple modes of action. 47 In tumor progression, epigenetic malfunctions display important roles in addition to genetic factors. An abnormal expression of EZH2 induces permanent silencing on some critical tumor suppressor genes, represses their transcription, and therefore contributes indirectly to tumor proliferation, invasion, and metastasis. Accordingly, it is highly expressed in several types of tumors such as breast cancer, prostate cancer, ovarian cancer, lymphoma, as well as lung cancer. 48 In NSCLC, overexpression of EZH2 mRNA was found to be a negative prognostic indicator and it was suggested as a biomarker to predict the response to histone deacetylase (HDAC) inhibitors. 49 On the contrary, EZH2 itself is an actionable molecular target and there are several ongoing clinical trials of EZH2 in hematological malignancies. 50 In a recent study, benzomorpholine derivatives were reported as novel EZH2 inhibitors for anti-NSCLC activity. 51 Perhaps EZH2 can be targeted molecularly with potent inhibitors in appropriate NSCLC patients for therapeutic purposes in the future. The next top hub gene found in our study, CDKN2A or p16INK4a, is a powerful inhibitor of CDK4/6 and capable of inducing cell cycle arrest in G1 and G2 phases. It acts as a tumor suppressor and its loss is a significant event in several cancer types. In the majority of the cases, CDKN2A is inactivated by homozygous deletions or through hypermethylation of the promoter region of the gene. However, in accordance with our data, a retained overexpression of CDKN2A may result in metastatic and invasive tumor phenotypes and is associated with poor prognosis. 52
CBL is a proto-oncogene associated with diseases such as juvenile myelomonocytic leukemia and other hematological cancers. 53 It acts as an E3 ubiquitin-protein ligase that can function as a negative regulator of many signaling pathways triggered by cell surface receptors such as FGFR1, FGFR2, PDGFRA, PDGFRB, and EGFR. 54 Although the role of CBL family proteins in NSCLC is largely unknown, in a few recent studies their upregulation were shown to inhibit especially mutated EGFR expression by mediating proteasome degradation of the protein. 55 Therefore, it may be speculated that the positive prognostic value of CBL in our study can be attributed to its overexpression and anticancer effects in patients with mutant EGFR expression.
Non-receptor tyrosine-protein kinase FYN is an oncogene that plays a role in several biological processes such as cell motility, cytoskeletal remodeling, cell adhesion, integrin-mediated signaling, and cell growth and survival. FYN is overexpressed in various cancers, including head and neck cancer, melanoma, squamous cell carcinoma, and prostate cancer. 56 High FYN expression was associated with epithelial-mesenchymal transition and has various roles in cancer-metastasis-related pathways. 57 On the contrary, the prognostic value of FYN was shown in a recent work aiming to predict metastasis in NSCLC. 58 Consistent with our results, they have shown a high FYN expression associated with an improved survival time. Although the reason is unknown, this contradiction with previous findings needs to be clarified by further studies in the future. One of the hub genes with a positive prognostic value in our PPI network analysis was LRKK2, which is a member of the leucine-rich repeat kinase family. LRKK2 is a serine/threonine kinase that modulates a large number of proteins involved in multiple processes like vesicle trafficking, autophagy, and neuronal plasticity. 59 Abnormalities in this gene are largely associated with neurodegenerative disorders such as Parkinson’s disease. 60 In a recent study, LRKK2 was shown to serve as a scaffold during activation of the WNT/β-catenin pathway, which is activated in different types of human cancers. 61 LRRK2 overexpression was shown to have important antitumor activities like suppression of proliferative, migrative, and invasive properties of tumor cells, and induction of apoptosis together with the arrest of the cell cycle. 62 LRRK2 was shown as a hub gene in colon cancer by a bioinformatics approach 63 and was found among frequently mutated genes in lung squamous carcinoma. 64 SOCS2 gene is a member of the suppressor of cytokine signaling family proteins and it takes part in the negative control of transduction in this signaling cascade through the Janus kinase (JAK)- signal transducer and activator of transcription (STAT) pathway and in part by ubiquitination. 65 In general, SOCS2 seems to act largely as a positive prognostic factor in different human cancers including hepatocellular carcinoma, breast cancer, and colorectal cancer, with an exception in acute myeloid leukemia (AML) in a large cohort. 66 Inhibition of SOCS2 with different miRNAs in lung cancer results in cell proliferation and metastasis. 67 In addition, SOCS2 was identified as one of the downregulated genes in lung tumor tissue in a cDNA array-based study. 68 Our survival analysis results have shown a longer patient survival time with high mRNA expression of SOCS2; therefore, it may be considered as a positive prognostic marker of NSCLC.
Overall, CDC20, AURKA, CDK1, EZH2, and LRKK2 were identified as important hub genes in other bioinformatics studies using various integrated analysis methods by utilizing some of the NSCLC datasets in our study. However, in the present study, we suggest CDKN2A, CBL, FYN, and SOCS2 as novel important biomarker candidates in NSCLC.
Our drug target analysis considering the identified top 10 DEGs has revealed 13 drugs or small molecules demonstrating a potential to reverse their aberrant expressions. Among these drugs, we identified Pracinostat, Trichostatin A (a histone deacetylase-HDAC inhibitor), S1169 (TGX-221-a PI3K inhibitor), HY-11001 (PHA-793887-a potent CDK inhibitor), AG-879, and IMD 0354 with antineoplastic effects. On the contrary, Z-Leu3-VS (a proteasome inhibitor), TGX-115 (an antimalarial agent), Ingenol 3,20-dibenzoate (an antiviral agent), Maprotiline hydrochloride (a selective noradrenaline reuptake inhibitor), NTNCB hydrochloride (a neuropeptide Y receptor Y5 antagonist), Rottlerin (antiplasmodial activity), and RF01079 with no functional information were also identified as potential drug candidates to reverse the expression of top 10 DEGs in our analysis. Pracinostat is a small-molecule HDAC inhibitor that may take part in chromatin remodeling through an accumulation of histone hyperacetylation. Combination therapy with pracinostat was suggested for elderly patients of AML in a phase II clinical trial. 69 However, we could not find any literature regarding the effects of pracinostat in NSCLC. Trichostatin A is another HDAC inhibitor that is identified in our study and is long known as an antineoplastic agent against NSCLC. 70 Although there are many reported studies on the antitumoral effects of Trichostatin A in NSCLC, perhaps the most critical impact was about the reversal of chemoresistance resulting from high IGFBP2 expression. 71 Trichostatin A was found to inhibit both EZH2 72 and AURKA activator FOXM1 73 which had an FC value above 2 and thus upregulated in all datasets we analyzed (Supplementary Table 1). As AURKA-induced CDK1 expression may trigger both CDC20 and EZH2 gene expressions, we propose an action mechanism in which Trichostatin A could inhibit all these hub genes through its negative actions on FOXM1 gene expression (Figure 6).

Proposed mechanism of action for reversing effects of Trichostatin A on hub gene expressions. Trichostatin A can interfere with AURKA expression through inhibiting FOXM1 activator. Reduced AURKA expression may interfere with CDK1 activation and thus expression of its downstream targets. Blocking EZH2 expression as well may help to decrease tumorigenic properties of NSCLC cells. Black arrow: downregulation. AURKA indicates Aurora Kinase A; CDK1, Cyclin Dependent Kinase 1; EZH2, Enhancer of Zeste 2 Polycomb Repressive Complex 2 Subunit; NSCLC, non-small cell lung cancer.
AG-879 or Tyrphostin AG-879 is a specific TKI for ErbB2 (HER2). Although its antineoplastic effects are long known, 74 the utility of AG-879 is very limited in recent cancer studies. IMD0354 is an IKK-2 inhibitor V with therapeutic effects on inflammation and insulin resistance. The antineoplastic effect of IMD 0354 was reported on breast cancer stem cells to prevent chemoresistance in a murine model. 75 As of now, its antitumoral effects and their molecular mechanisms in NSCLC largely remain to be elucidated. In a recent study, it was shown that IMD0354 could effectively suppress cancer cell proliferation, invasion, and migration through inhibition of elevated expressions of transmembrane serine protease 4 (TMPRSS4) in NSCLC cells. 76 Therefore, IMD0345 has considerable potential for further development as a novel anticancer agent for NSCLC treatment. S1169 or TGX-221 is a potent PI3K isoform-specific inhibitor that was derived from a natural compound Quercetin. 77 The derivatives of TGX-221 have been utilized especially against prostate cancer cells as well as in xenograft models. 78 Antitumoral effects of TGX-221 in NSCLC are somewhat contradictory and the number of studies is limited. It was suggested that PI3K isoforms may functionally compensate for one another thus limiting the efficacy of single-agent treatment. 79 HY-11001 or PHA-793887 is a potent, ATP-competitive CDK inhibitor acting on CDK1, CDK2, CDK4, and CDK9, which has been used in a phase I clinical trial studying the treatment of advanced or metastatic solid tumors. 80 However, this study was not continued due to severe hepatic toxicity. There was no evidence for in vitro or in vivo antitumoral effects of PHA-793887 in NSCLC. Although there is a line of evidence for drug repurposing for other antimalarial drugs such as hyroxychloroquine and artemisinin, 81 there was no indication of TGX-115 usage in NSCLC treatment. We could not find any cancer-associated literature for remaining small molecules Z-Leu3-VS, TGX-115, Ingenol 3,20-dibenzoate, Maprotiline hydrochloride, NTNCB hydrochloride, or Rottlerin. On the contrary, RF01079 was found to be inactive in most of the tested bioassays according to PubChem. Overall, we identified Trichostatin A, IMD0354, and TGX-221 as potent drug repurposing candidates for NSCLC treatment in our study.
This study integrates gene expression profiles and PPI networks to identify prognostic genes and candidate drug molecules. This analysis, however, is limited in that it does not include the expression of these genes in the different NSCLC subtypes. Here, we showed that high expressions of CDC20, AURKA, CDK1, EZH2, and CDKN2A genes were associated with significantly poorer overall survival, whereas upregulations of CBL, FYN, LRKK2, and SOCS2 were associated with a significantly better prognosis. Our drug target analysis of hub genes suggests a potential use of Trichostatin A, Pracinostat, TGX-221, PHA-793887, AG-879, and IMD0354 antineoplastic agents to reverse the expression of hub genes in NSCLC patients. While our work showed the benefit and usefulness of gene expression data analysis in bringing out the DEGs and hub genes that can be potential prognostic and treatment targets of NSCLC, the need for improved analysis and prospective clinical studies is still imperative. Finally, this study can contribute to the overall understanding of the underlying molecular mechanisms of NSCLC and serve as a guide to subsequent experimental studies.
Supplemental Material
sj-xlsx-1-bbi-10.1177_11779322221088796 – Supplemental material for Integrative Analysis for Identification of Therapeutic Targets and Prognostic Signatures in Non-Small Cell Lung Cancer
Supplemental material, sj-xlsx-1-bbi-10.1177_11779322221088796 for Integrative Analysis for Identification of Therapeutic Targets and Prognostic Signatures in Non-Small Cell Lung Cancer by Özgür Cem Erkin, Betül Cömertpay and Esra Göv in Bioinformatics and Biology Insights
Footnotes
Author Contributions
ÖCE, EG study conception and design. ÖCE data collection. ÖCE, BC analysis and interpretation of results. ÖCE, BC and EG draft manuscript preparation. All authors reviewed the results and approved the final version of the manuscript.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.