
Abstract
We present an approach for detecting enzymes that are specific of Leishmania major compared with Homo sapiens and provide targets that may assist research in drug development. This approach is based on traditional techniques of sequence homology comparison by similarity search and Markov modeling; it integrates the characterization of enzymatic functionality, secondary and tertiary protein structures, protein domain architecture, and metabolic environment. From 67 enzymes represented by 42 enzymatic activities classified by AnEnPi (Analogous Enzymes Pipeline) as specific for L major compared with H sapiens, only 40 (23 Enzyme Commission [EC] numbers) could actually be considered as strictly specific of L major and 27 enzymes (19 EC numbers) were disregarded for having ambiguous homologies or analogies with H sapiens. Among the 40 strictly specific enzymes, we identified sterol 24-C-methyltransferase, pyruvate phosphate dikinase, trypanothione synthetase, and RNA-editing ligase as 4 essential enzymes for L major that may serve as targets for drug development.
Introduction
Leishmaniasis is a disease caused by a protozoan parasite from more than 20 species in the Leishmania genus of the family Trypanosomatidae that may affect skin (cutaneous form) and the mucosal membranes (visceral form); the visceral form can be fatal. Trypanosomatidae is successively inserted in the order Kinetoplastida, the subclass Metakinetoplastina, the class Kinetoplastea, the phylum Euglenozoa and the clade Excavates. The parasite is transmitted by blood-sucking insects known as sandflies, a colloquial name for any species or genus of flying, biting, blood-sucking dipteran encountered in sandy areas. In the New World, leishmaniasis is spread by phlebotomine (Diptera, Psychodidae, Phlebotominae) of the genus Lutzomyia, 1 whereas in the Old World, it is spread by sandflies of the genus Phlebotomus. 2 The World Health Organization (WHO) estimates that there are 0.7 to 1.3 million new cases worldwide3,4 with 20 000 to 30 000 deaths per year. Medications include the following: (1) liposomal amphotericin B, a combination of pentavalent antimonials and paromomycin, and miltefosine, for the visceral form and (2) paromomycin, fluconazole, or pentamidine, for the cutaneous form. 5
Briefly, (1) liposomal amphotericin B is thought to act by binding to ergosterol, the principal sterol in fungal and Leishmania cell membranes, 6 which results in a change in membrane permeability promoting monovalent ion leaks, metabolic disturbance, and cell death. AmBisome, the commercial formulation, is expensive, which makes its large-scale use in developing countries problematic. (2) The effect of pentavalent antimonials is pleiotropic because it may act (a) as a prodrug that is converted to active and more toxic trivalent antimony, (b) directly on molecular targets in the thiol redox metabolism 7 such as trypanothione and glutathione, 8 and (c) through thiols and ribonucleosides. 9 (3) Paromomycin is an antibiotic that was proposed to successively (a) bind to the paraflagellar rod proteins and prohibitin, (b) be internalized by endocytosis, and (c) interact with a P-type H+ adenosine triphosphatase (ATPase). 10 (4) Miltefosine is an alkyl phospholipid whose mechanism of action is to inhibit phospholipid metabolism by decreasing phosphatidylcholine and increasing phosphotidylethanolamine 11 with the consequence of apoptosis-like cell death. 12
Amphotericin B, pentavalent antimonials, paromomycin, and miltefosine induce noxious side effects and have variable efficacy in leishmaniasis treatment depending on the geographical locality. A combination treatment has the potential advantages of shortening the duration of treatment, reducing the overall dose of medicines, and reducing the probability of selection of drug-resistant parasites. Several trials of combinations have been conducted, with favorable results. 13
In the case of the cutaneous form fluconazole and pentamidine may also be part of a combination. 13 The New World form of Leishmania tends to be more severe and lasts longer than that of the Old World form. No single treatment approach fits all possible clinical presentations. In fungi, fluconazole interacts with 14-α demethylase, a cytochrome P450 enzyme necessary to convert lanosterol to ergosterol. As ergosterol is an essential component of the fungal cell membrane, inhibition of its synthesis results in increased cellular permeability, causing leakage of the cellular contents 14 ; in Leishmania, the precise mechanism of action has not been described yet. 15 Interestingly, it is well tolerated at relatively high doses by humans. 16 Pentamidine was found to be a competitive inhibitor of arginine transport 17 and a noncompetitive inhibitor of putrescine and spermidine transport in Leishmania infantum, 18 Leishmania donovani, and Leishmania mexicana. 19
Infections caused by Leishmania are becoming major public health problems on a global scale. Many species of Leishmania around the world are obtaining up to 15-fold resistance levels, as estimated by the WHO. The arsenal of drugs available for treating Leishmania infections is limited and includes pentavalent antimonials, pentamidine, amphotericin B, miltefosine, fluconazole, and a few other drugs at various stages of their development process. 20 Leishmania that is showing resistance is relatively difficult to observe and maintain in laboratory settings. 21
The pharmaceutical industry has experienced a dramatic decrease in productivity between the 1980s and 2010 that is principally due to the cost burden of investing in the research and development (R&D) of new drugs, 22 which are estimated to cost $1.7 billion each. 23 In response, the process of drug R&D is shifting from how these activities were addressed as health care priorities in the past to approaches that are dominated by their potential market value. Even if the situation reverted to the figure of the 1980s (as defined by the number of new chemicals licensed by the Food and Drug Administration), concerns exist regarding future decision making, which requires a new paradigm for the management of R&D activities to attend to global needs. 24
Of course, screening for active compounds must be continued to anticipate the development of resistant parasitic forms. However, due to its low market potential, R&D for drugs against parasitic diseases that are endemic to tropical regions in developing countries is generally under prioritized or neglected by private companies and its realization depends on other means.25,26 As alternative financial sources for research on neglected diseases are limited and highly divided among individual researchers, open science and data sharing have received a growing interest as means of leveraging and combining the available resources to accelerate drug discovery efforts. 27 This community-based concept for a new drug discovery model led to the London Declaration on Neglected Tropical Diseases in 2012 for the control, elimination, or eradication of neglected tropical diseases (http://unitingtocombatntds.org).
Many proteins are potential targets for drug interventions that control human diseases. The most recent number of drug targets was estimated to be in the hundreds, based on an analysis made before 2007. 28 However, the number of druggable proteins is substantially greater according to the DrugBank database Web site (http://www.drugbank.ca/). The current version of this database (5.0) contains 8206 drug entries that are linked to 4333 nonredundant (nr) protein sequences (ie, drug target/enzyme/transporter/carrier).
The discovery of drugs based on in silico docking of the inhibitors in models of the 3-dimensional (3D) structures of the protein targets has proven to be of great value in the processes of rational drug design as well as drug screening and has effectively contributed to conserving resources in the area of drug discovery. The use of this information may allow a substantial savings in the cost and time involved in the process of drug release. 29
Unfortunately, all druggable protein targets are not necessarily suitable for a therapeutic design because of several constraints, including noxious side effects to patients. The specific inactivation of a pathogenic enzyme without affecting any human enzymes would provide a safe approach to control the pathogen. The long-lasting, diverging evolution over a billion years that separates the common origins of simple unicellular and complex multicellular eukaryotes (phytozoan and metazoan) could have given rise to the enzymatic functions that are specific to parasitic pathogens in comparison with their hosts. 30 Similar mechanisms could generate analogous enzymes in pathogens and hosts that can also provide potential molecular targets due to the variations in their enzymatic sites that can be distinguished for inactivation by a given inhibitor. 29 By definition, analogous enzymes result from the convergent evolution of independent proteins rather than originating from common ancestral proteins (homologs), which allows for the same function but have differences in their primary, secondary, and tertiary structures. 31
A previous study established a methodology to identify, annotate, compare, and study analogous and homologous enzymes. The results obtained with this method could be used to identify the enzymatic activities in essential metabolic pathways that can serve as new therapeutic targets for fighting the infectious and parasitic diseases caused by protozoa of the Trypanosoma and Leishmania genera. 32 This methodology includes the computational search for genes that encode (1) enzymes that are specific to parasites and, thus, are not encoded in human DNA, (2) analogous enzymes, and (3) the in silico mapping of the biological pathways of analogous and homologous enzymes considering the entire metabolism of the pathogens under consideration. The resulting pipeline, called AnEnPi (Analogous Enzymes Pipeline, http://anenpi.fiocruz.br), uses the protein sequences stored in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg/). AnEnPi implements algorithms that can perform the following tasks: (1) the clustering of protein sequences by homology using Basic Local Alignment Search Tool (BLAST); (2) the classification of sequences in different homologous clusters as analogous enzymes when they have the same enzymatic function, ie, Enzyme Commission (EC) number 33 ; (3) the detection of specific enzymes 34 ; (4) the annotation of protein functions using BLAST or HMMER; and (5) the generation of metabolic maps using the tools provided by KEGG (http://www.genome.jp/kegg/tool/map_pathway1.html). The reconstruction of metabolic pathways in parasites based on the metabolic maps provided by KEGG aims to identify the enzymatic activities that are essential for a parasite and can be considered promising targets for drug development.35–37
Any error associated with a classification process can be qualified as follows: (1) false positives, which occur when a Leishmania major enzyme is classified as specific or analogous while at least 1 gene for a homologous enzyme exists in the Homo sapiens genome; (2) false negatives, which occur when the classification of a homologous pair is erroneous, and it should be classified as a specific or an analogous enzyme because of an incorrect annotation; (3) true positives, which occur when a pair of specific or analogous genes is correctly classified; and (4) true negatives, which occur when a pair of homologous genes indeed does not refer to specific or analogous genes.
The occurrence of false positives based on the available sequence annotations can result in significant errors and has been a recurring problem in the previously described methodology. Often, incomplete annotations are responsible for generating the false positives of specific or analogous enzymes. Because annotation is a dynamic process that improves with time, at any given moment, a pair of enzyme-encoding genes can be incorrectly classified as specific simply because the homologous counterpart in 1 genome of the pair has not yet been annotated.
Here, this report focuses on an evaluation of the false positives produced by AnEnPi because of the following: (1) in its current iteration, AnEnPi does not have any process of false positive assessment and (2) this error component is critical for decisions on the investment of limited resources on a putative specific or analogous enzyme for drug development. The existence of false positives during target identification could invalidate years of research into drug development. We only considered the errors resulting from incomplete annotations here, not those from mistakes in the genome assemblies or EC annotations, and we describe a method to track false positives in the AnEnPi output to improve the identification of enzymes that are specific to L major compared with H sapiens.
As a result of the application of the proposed depuration process to L major, we found 4 specific enzymes (sterol 24-C-methyltransferase, RNA-editing ligase, pyruvate phosphate dikinase, and trypanothione synthetase) that seem to be valuable targets for further drug development.
Materials and Methods
Background
AnEnPi has produced false positive diagnoses due to inaccurate and fragmented annotations. Here, we propose a process to debug these error sources. We searched for false positives in a list of enzyme sequences obtained from AnEnPi 32 whose functions were annotated as specific for L major compared with H sapiens. As a pipeline, AnEnPi classifies enzymes as homologous, analogous, and specific enzymes by considering the similarities among sequences with ⩾100 amino acids and the functional annotations of the enzymes in the 2 organisms being compared. Considering all the combination pairs of enzymes between 2 organisms, an enzyme was designated (1) homologous to another when the similarity score and the E-value of their alignment was, respectively, ⩾120 and close to 10−4 or below (using BLASTp); (2) analogous to another when the similarity score of their alignment was <120, but the enzymes were associated with the same EC (catalyzing the same reaction); (3) functionally specific when it was homologous to another, as defined under “1” but associated with a different EC; and (4) strictly specific when it was not homologous and did not share its EC with any of the other enzymes. In other words, by classifying enzymes as strictly specific to L major, we meant that the reactions of these enzymes were found to be catalyzed in L major but not in H sapiens. Thus, an enzyme was considered strictly specific to L major when its EC was not found in H sapiens. The gene list that we recovered from AnEnPi (KEGG release 58.1, June 1, 2011) included 67 sequences from L major, with 42 of these sequences being associated with a respective EC that was classified as strictly specific to L major compared with H sapiens by AnEnPi.
Process of strictly specific enzyme depuration
We could distinguish several steps and components in the process of strictly specific enzyme depuration. First, it is necessary to diagnose whether or not the putative strictly specific enzymes of L major have homology with human DNA and its encoded enzymes. Second, a human subject enzyme that was found to be a homolog might be associated with the same EC as the L major query and could be diagnosed as a false positive or it could be associated with a different EC number and be diagnosed as functionally specific. Third, in the case of an ambiguous situation, we also characterized the 2-dimensional (2D) structure of the homologous regions. Fourth, we further checked putative strictly and functionally specific enzymes with 3D-HMM (HHpred and HHsearch) to detect eventual remote homologies in reference to the human 3D enzyme structures. Fifth, the enriched set of strictly specific enzymes of L major was challenged for the potentiality of its components as suitable as lead targets in reference to their centrality in metabolic pathways. Sixth, specific enzymes were investigated in other human parasites to assess their relevance as targets for drug development.
The process that we followed to enrich our enzyme set in the strictly specific proteins of L major that are potentially suitable for drug development is summarized in Figure 1 and is as follows. (1) The comparison of L major DNA-encoding proteins with human DNA was straightforward and it gave the sequence coordinates where homologous sequences could be found in the human genome (Figure 1A). The consistency of the detected homologies could be analyzed by performing multiple comparisons to determine whether the corresponding matches were obtained from protein-to-protein and protein-to-DNA using parasite-to-human and human-to-parasite queries (Figure 1B). (2) The amino acid sequence comparison between L major enzymes and human proteins allowed us to quickly identify most of the human proteins that deserved attention as potential homologous enzymes (Figure 1C). (3) The comparison of the human proteins identified under “2” with human chromosomal DNA allowed the delineation of their gene structure (exons and introns). (4) The comparison of the L major proteins that had a homologous hit with the human genomic sequence without a corresponding human protein in the Ensembl list 38 indicated a potentially missing human gene annotation or a possible pseudogene (Figure 1D). (5) The subject enzyme of a homologous pair is associated with an inconsistent annotation (Figure 1E) or a different EC compared with the query enzyme (Figure 1F). (6) We compared the 2D profiles of the query and subject sequences in ambiguous homologous pairs to discriminate orthology and distant paralogy or analogy (Figure 1G). (7) Ambiguous annotations, as in “6,” also occurred when an enzyme that was classified as nonhomologous to the human genome was found to share an EC with that of a homologous one in L major, which is a case of intragenomic analogy. In that case, the nonhomologous L major enzyme might be analogous to the human counterpart of the homologous one, as it may occur when the EC of the human counterpart was incomplete or absent (Figure 1H). (8) We challenged the putative strictly and functionally specific enzymes of L major for remote homologies with human enzymes using HHpred and HHsearch (Figure 1I). (9) We challenged the putative strictly specific enzymes of L major for their essentiality in KEGG metabolic pathways (Figure 1J). (10) To assess the relevance of the proposed strictly specific enzymes of L major for drug development, we analyzed the distribution of these enzymes in human parasites (Figure 1K).
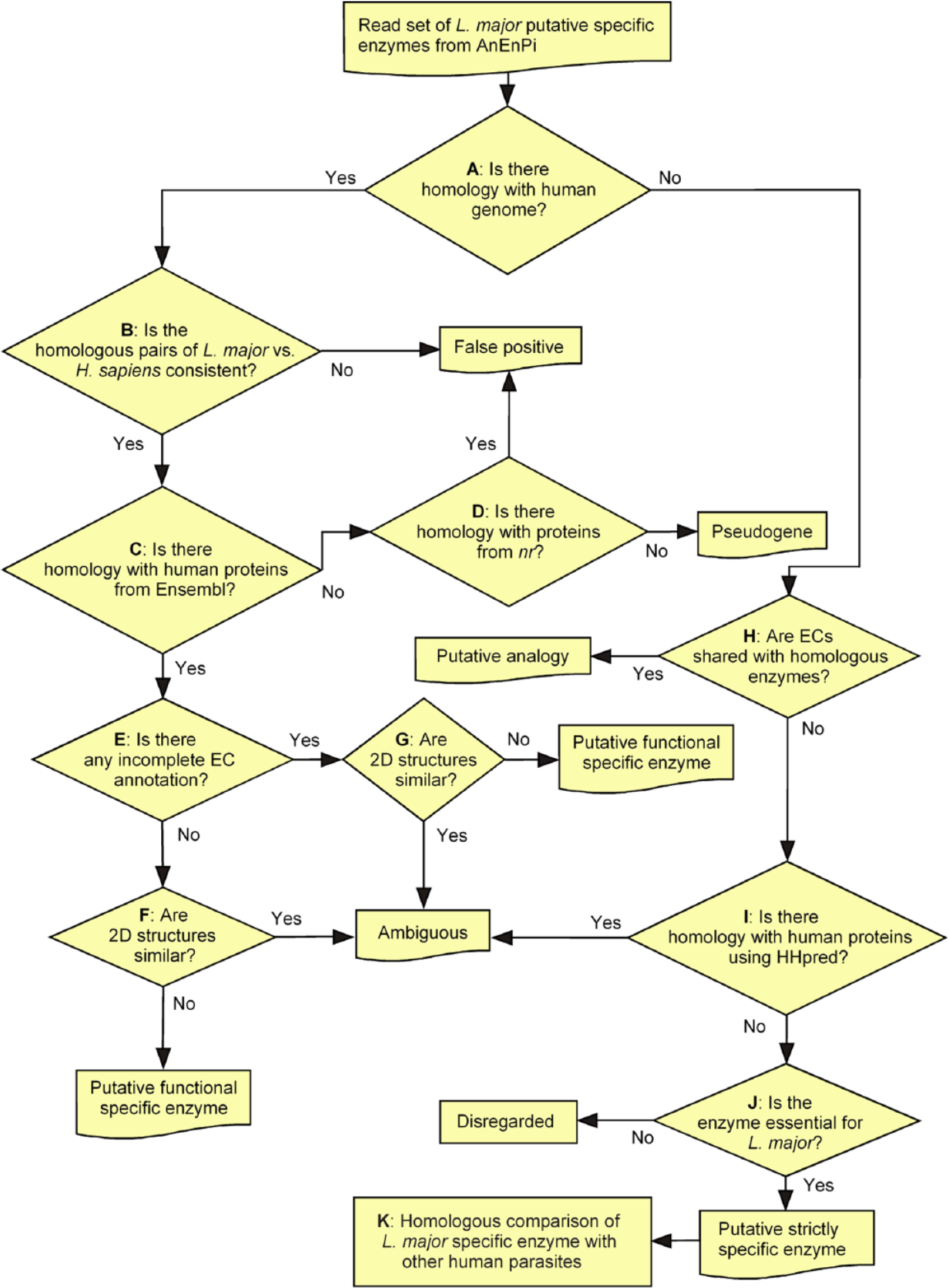
Flowchart of strictly specific enzyme depuration.
Mapping Ensembl and L major homologous proteins on the human genome
To determine whether the classification of specific enzymes might produce false positives, we first searched for homology between the DNA-encoded proteins of L major and the DNA sequence of the human genome (Figure 1A). To this end, we employed the sequences from the putative L major–specific proteins available from TriTrypDB (release 6.0—September 2013) 39 and compared them with the human genome sequence (Ensembl, release 74—November 2013) using tBLASTn. We considered a pair to be a consistent homologous hit when the E-value of a tBLASTn alignment was ⩽10−4 and its score value was ⩾120. When consistent hits were obtained, the genomic coordinates from the tBLASTn output were recovered and compared with the genomic coordinates of the encoded protein sequences available from Ensembl (release 74—November 2013) to determine whether a protein annotation might exist for the genomic region corresponding to the tBLASTn hit (Figure 2).

Homologous hit search algorithm for the classification of false positives of parasite-specific genes in humans. (A) The 11 possibilities of Ensembl protein associations (S2-E2) that one may obtain with a human genomic region that is homologous (tBLASTn) to a parasite protein query (S1-E1). S and E are for the beginning (start) and end of a tBLASTn homology or human gene coordinates. The Boolean description of each association between a tBLASTn hit in a human genome region and the Ensembl proteins annotated in that region is given on the left and right sides of panel A. “&” is used here in its Boolean sense, ie, a logical AND. Human genes for the Ensembl protein that are eventually compatible with a parasite’s homologous counterpart (tBLASTn) are modeled by thin lines. Human genes for Ensembl proteins in the same genomic region as a parasite’s homologous counterpart, but which are not compatible with it, are represented by dashed lines. (B): The decision tree for TRUE and FALSE associations of the human genes for the Ensembl proteins with a human genomic region that had a homology (tBLASTn) hit with a parasite protein.
Thinking of the automation of that process, we identified 11 possibilities (Figure 2A), of which 7 are considered TRUE and 4 are considered FALSE in the Boolean sense. The FALSE and TRUE options can be easily diagnosed according to the decision tree shown in Figure 2B.
Consistency in homologous pair of L major vs H sapiens using BLAST
The lack of introns in L major genes may complicate the direct comparison of their gene sequences with those of the human genome, as the homologous regions, if any, could be interrupted by a splicing site in the human sequence of the homologous gene. To clarify this type of ambiguity, we checked whether a genomic hit obtained by comparing the L major proteins with the DNA sequence of human chromosomes (tBLASTn), as described in the previous paragraph, also matched the homologous region detected in the BLASTp (PSI-BLAST [Position-Specific Iterative BLAST]) comparison of the same L major query with the human protein sequence (subject) corresponding to that genomic hit. Thus, when an Ensembl protein annotation for a homologous hit of a L major protein with the human genome did exist (Figure 1B, Figure 3A), it was checked for consistency with the gene model, as found by tBLASTn comparison (Figure 3B) and from National Center for Biotechnology Information consensus coding sequence (CCDS)40,41 (Figure 3C), and, if consistent, it (them) was (were) assigned to the considered genomic region where its homology with the L major enzyme was obtained.
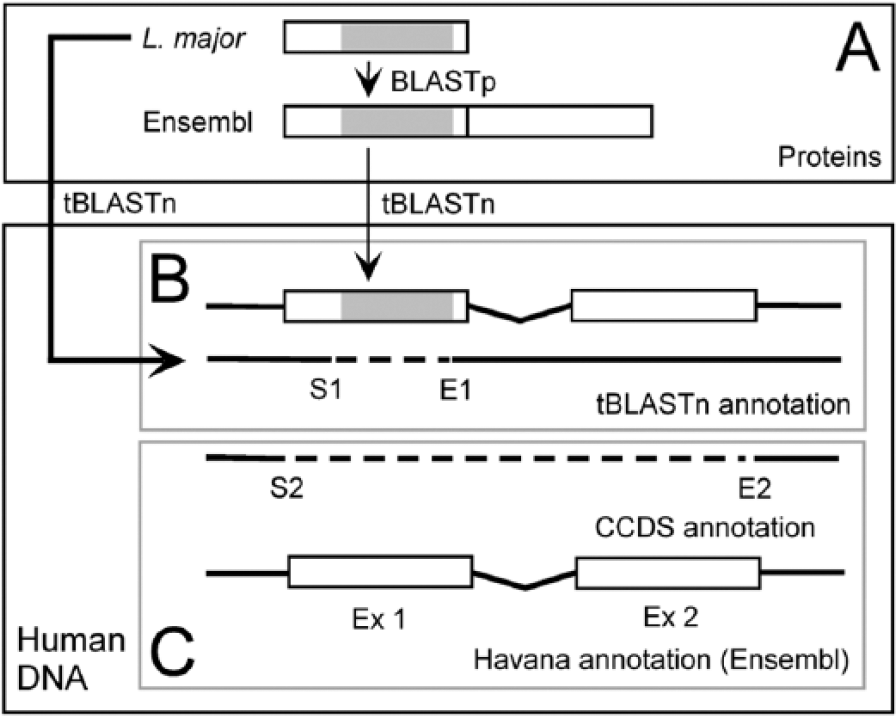
Search scheme for false positives of parasite-specific genes in human. The (A) homologous hit of Leishmania major and human proteins is compared with (B) the human gene structure obtained by tBLASTn search and with (C) the CCDS annotation from Havana.
To map a homologous region of a L major protein on its tagged human gene (Figures 1A and 3), we must consider several lines of evidence. (1) Homology may be split into 2 homologous regions in the subject sequence if an intron falls in its genomic region corresponding to the protein domain of the query that is involved in the homology. In this case, the 2 subject homologies would be contiguous in the query regarding the final coordinate of the first region and the initial coordinate of the second region. (2) The genomic coordinate of the final position of a homologous region minus the genomic coordinate of its initial position plus 1 must be a multiple of 3 because of the codon structure of CCDSs. (3) The homologous region encodes a protein, and thus, the subject coordinates must match exons (Figures 1B and 3B).
After comparing the L major proteins with human proteins (BLASTp) in Ensembl (Figures 1C and 3A), we may consider several steps to automate the process of homologous region mapping to human exons (Figure 3). (1) Considering the significant match of this comparison, we recovered the coordinates of the human gene according to the chromosome sequences (Figure 3B). (2) To determine the gene model corresponding to a human protein that has a significant hit with L major proteins, we compared (tBLASTn) that human protein from Ensembl to its respective DNA sequence stretch by extracting it with the chromosomal coordinates of the corresponding initial and final gene position as informed by the Ensembl annotation (Figure 3C). (3) The homologies detected by the comparison of an Ensembl protein with its corresponding human DNA stretch, as described under “3,” gave us the coordinates for the exons (subject) relative to a gene model. (4) To translate the exon coordinates under “3” into coordinates relative to the chromosome context, we added the exon coordinates to the initial position of the corresponding gene in chromosomal coordinates as provided by Ensembl. (5) To calculate the intron size, we subtracted the chromosomal coordinate (under “4”) of the beginning of an exon from the chromosomal coordinate of the end of the preceding exon for each exon of the corresponding gene. (6) To calculate the exon size, we subtracted the chromosomal coordinate (under “4”) for the end of an exon from the chromosomal coordinate for its beginning and added 1; we did this for all exons of each gene. Of course, the size of all the exons had to be a multiple of 3 (codons) to be considered relevant. (7) The sum of the exon size divided by 3 had to give us the protein size, which was inferred from the model obtained from the chromosomal sequence. The protein size could then be compared with the size of the query protein from Ensembl for size consistency. (8) Another consistency test was performed by determining whether the strands (“+” or “−”) from the exons, which were deduced from the subject homologies, were all “+” or all “−.” Exons in alternating “+” and “−” strands for the same gene with homology to the same query protein would not make sense, and this result allowed us to diagnose cases of inconsistency. (9) We also confirmed the gene models that we obtained from the tBLASTn search of the Ensembl proteins with human DNA by comparing the hit coordinates with the exon coordinates available in the CCDS database, as annotated by Ensembl. (10) To map the homologous region of a L major query with a human exon, we subtracted the initial and final chromosomal (human) coordinates (the subject) that corresponded to the hit with the initial and final coordinates of each human exon (obtained in “4”), respectively. The exon for which both subtractions gave values equal to 0 (or close to 0 providing that the small protruding end was a multiple of 3 bases) was considered the one that matched the homologous region in the L major query.
We considered the homologies between a L major protein and a human protein in the Ensembl list to be true positives when (1) the human protein was legitimate (all exons mapped on the same DNA strand) and (2) the homologous region between both proteins (from H sapiens and L major) matched the coding frame and coordinates of the genomic sequence that corresponded to the hit of the comparison (tBLASTn) of the L major proteins with the chromosomal DNA. In other words, to be considered significant, a homology between L major and a human protein had to be consistent with the gene model of that human protein, ie, the homologous regions between the proteins and DNA had to match the exons (Figures 1B and C and 3). All these operations were gathered together in a single Excel spreadsheet that we used as a dashboard (see Table S1).
Genomic hits without counterparts in Ensembl proteins
In cases where a genomic hit obtained by tBLASTn did not match the position of any human protein in the Ensembl list (Figure 1D), it was considered as an indication that the human gene had not received a designation during the annotation process by Ensembl. The absence of an annotation was addressed by translating the homologous stretch of the human genome (subject) into amino acids and using the protein sequence for a homology search (BLASTp) with the nr section of GenBank (release 201.0—April 2014). When a human homologous protein was found in nr, its complete sequence was compared (tBLASTn) with a DNA stretch of 20 kb around the initial L major hit with the human genome to find the gene model (if any) associated with this protein.
The particular case of functional specificity
Considering the instances of enzymatic functional specificity between the 2 homologous proteins of L major and H sapiens in which the proteins were annotated as having differences in their enzymatic functionality (based on the assigned EC), we distinguished between the following categories (Figure 1E, F and H): (1) Uncertainty with the annotations occurred when at least one of the enzymatic functions in both proteins could not be confirmed (incomplete or unavailable EC). In these cases, we investigated the protein’s name and function in UniProtKB/SwissProt 42 and attempted to verify whether the function described in this database matched that of TriTrypDB. If the function of the enzymes under comparison was synonymous in both organisms, the L major protein was diagnosed as a false positive for functional enzymatic specificity regarding its human counterpart; otherwise, functional specificity was assigned to the L major enzyme. This conservative position was taken because, if the members of a homologous pair had the same function, the potential of the negative collateral effects on the host of an inhibitor against the parasite’s enzyme would be greater if both the host and the parasite’s enzymes are orthologs. (2) Putative enzymatic functional specificity occurred in cases where the difference in enzymatic function of the homologous proteins could be confirmed. When both genes in the pair are paralogs rather than orthologs, they have an increased likelihood of having some sort of functional enzymatic specificity. 11 However, an inhibitor designed against the L major target may still affect the paralogous host counterpart at a lower rate.
Protein domain mapping and secondary structure
To better characterize whether ambiguous enzyme annotations (Figure 1G) were derived from distant paralogous or analogous enzymes, we searched their catalytic cores by mapping the domain composition for the conserved regions of the H sapiens and L major homologous pairs using PROSITE, Pfam, 43 and the Conserved Domain Database 44 for homology comparison with identity and score rates of ~40% and ⩾120, respectively, and predicted their secondary structure (2D) profiles using PRALINE. 45
Remote homology detection with hidden Markov models
In addition to BLAST, we used HHpred (http://toolkit.tuebingen.mpg.de/hhpred)46,47 and HHsearch (http://mobyle.rpbs.univ-paris-diderot.fr/cgi-bin/portal.py), 48 which implement hidden Markov models (HMMs) for a pairwise comparison of the profiles from sequence alignments in databases (Figure 1I). HHpred detects the homologous sequences in humans with a higher sensitivity than BLAST. HHpred was performed with the default options, ie, local alignment and the scoring of secondary structure similarity, and HHsearch was executed with the Protein Data Bank (PDB; http://www.rcsb.org/pdb/home/home.do), HHblits (as the alignment generation method), 49 85% (as sequence coverage), 10−6 (P value), and 10 (as maximal hit number). The sequences resulting from the first BLAST screening were further analyzed through HHpred and HHsearch to identify the possible remote sequence homologies in the human proteome that could result in false positives for the L major–specific enzymes. For a 3D model obtained by homology comparison to be consistent, a minimum identity level of 35% over 85% of the sequence template (subject) is required. Model consistency is necessary to evaluate the similarity between a query and its subject at the 3D level 50 to be able to diagnose whether both sequences are homologous or not. Alignments based on the primary sequences obtained from BLASTp may not have the necessary detection sensitivity and accuracy 49 to identify a homologous pair for the similarity levels at the border of the twilight zone. 51 Thus, the comparatively higher sensitivity and accuracy of HHpred and HHsearch 46 is another filtering step that is necessary for detecting false positives in the putative strictly specific enzymes of L major.
Metabolic pathway significance
To clarify the relevance of the putative enzyme targets of L major in terms of the metabolic impairment that results from their inactivation, we searched their respective EC in the KEGG pathway database (http://www.genome.jp/kegg/pathway.html). Then, we searched their metabolic pathway insertion to diagnose whether their inactivation may potentially affect L major without having deleterious consequences on human metabolism and, consequently, whether these enzymes can be used as targets for drug development (Figure 1J).
Sequence comparison of strictly specific enzymes from L major with other parasites
Because an enzyme that was specific to L major compared with H sapiens could also have homologous pairs in other human parasites (Figure 1K) and eventually serve as a target for drug development in these organisms, we searched for sequence homologies between the strictly specific enzymes of L major and the proteins in the organisms listed in Table 1.
Sequence materials of some important human parasites for comparison with the strictly specific enzymes of Leishmania major.
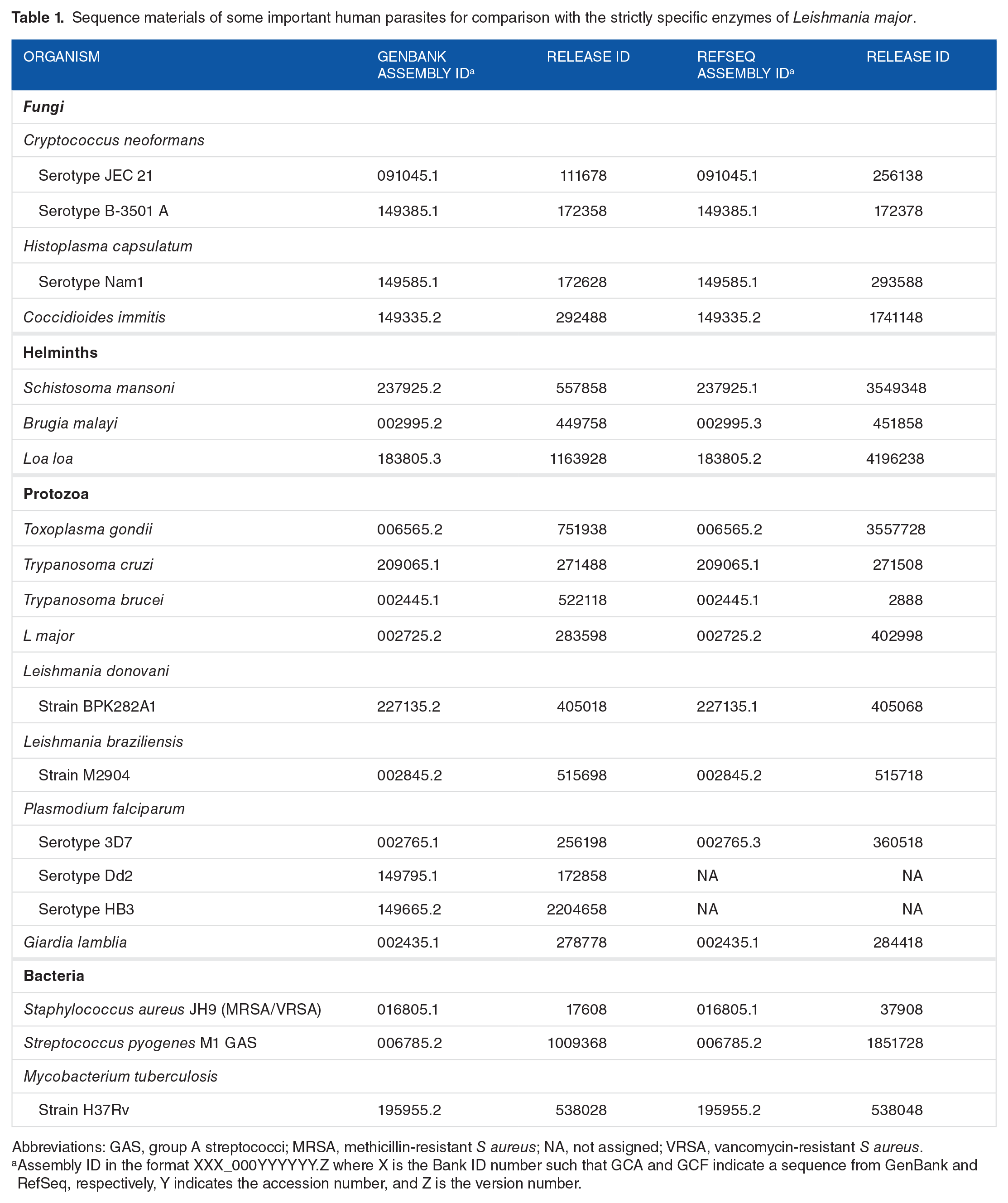
Abbreviations: GAS, group A streptococci; MRSA, methicillin-resistant S aureus; NA, not assigned; VRSA, vancomycin-resistant S aureus.
Assembly ID in the format XXX_000YYYYYY.Z where X is the Bank ID number such that GCA and GCF indicate a sequence from GenBank and RefSeq, respectively, Y indicates the accession number, and Z is the version number.
When an enzyme from these organisms was found to be present in KEGG (release 81.0—January 1, 2017) by comparing their EC annotation with that of L major, we acknowledged it. When it was not present in an annotation comparison, we proceeded with a sequence comparison using BLASTp. We acknowledged the homologies with the L major query as significant when the homology region was at least 75% of the query and the identity was ⩾35%, had an E-value ⩽10−4, and a score ⩾120. 50
Phylogenetic analysis
On one hand, the lethal effect of REL1 inhibition has been shown in Trypanosoma brucei by Panigrahi et al, 52 who identified MP52 as the sequence associated with REL1. 53 The association of REL1 sequences between T brucei, Trypanosoma cruzi, and L major has been clearly established by alignment 54 and allowed us to unambiguously assign the REL1 function to LmjF.01.0590. On the other hand, complete genome sequences were produced for Bodo saltans and Trypanoplasma borreli that belong, respectively, to 2 different orders, ie, Eubodonida and Parabodonida, within Metakinetoplastina. 55 This relation allowed us to test the hypothesis of whether the REL sequences of EC 6.5.1.3 emerged only within Trypanosomatidae or at the higher taxonomical level of Metakinetoplastina.
From the complete genome sequences of B saltans (GenBank Assembly ID: GCA_001460835.1, release ID: 2707578) and T borreli (http://www.sanger.ac.uk/resources/downloads/protozoa/trypanoplasma-borreli.html), we found the sequences encoding REL1 within the sequence NODE_50574 through a tBLASTn search of LmjF.01.0590 in the file Trypanoplasma_borreli_contigs_20120411.fa.gz.
We aligned the REL1 sequences of T brucei, T cruzi, L major, B saltans, and T borreli with ClustalW 56 and eliminated the column corresponding to gaps, and we searched the best phylogenetic relationship using maximum likelihood with MEGA7. 57 The substitution model was Jones-Taylor-Thornton, the rate among sites was considered uniform, and the tree inference was obtained by nearest neighbor interchange.
Results
Filtering pipeline of strictly specific enzymes of L major compared with H sapiens
Comparing the complete sets of (1) 8265 sequences of L major with 1547 that are annotated with ECs (341 nr ECs) and (2) 20 834 sequences of H sapiens with 6599 that are annotated with ECs (384 nr ECs) from KEGG, AnEnPi released a list of 67 protein sequences corresponding to 42 enzymatic activities that were putatively specific to L major compared with humans.
From the 67 enzymes (42 ECs) released by AnEnPi, 15 sequences (13 ECs) from L major produced 79 homologous hits with the human genome in CCDSs, as annotated by Ensembl, leaving 52 L major enzymes (29 ECs) without any homologies in the human genome (Figure 4A). By comparing the coordinates of the gene structures (exons + introns) reported by Ensembl for these CCDSs with the coordinates of the homologies given by the tBLASTn search of their protein sequence with the human genome (putative exons), we identified many inconsistencies in the Ensembl CCDS annotations (Figure 4B). By analyzing the 79 hits, we identified 64 nonconsistent CCDSs where the putative exons were (1) associated with the same protein, but they appeared on both gene strands, which is impossible given the translation process or (2) simply deprived of a protein association, making any further inference impossible (see Table S1).

Flowchart of the sequence depuration process in Leishmania major compared with Homo sapiens. Nrd stands for nonredundant.
Among the 15 L major enzymes, 14 demonstrated homology to the H sapiens proteins from Ensembl and 1 (LmjF.23.0270, pteridine reductase 1, EC 1.5.1.33) had a hit with the human genome sequence itself, but not with the CCDSs annotated by Ensembl (Table 2). Table 2 shows that in addition to LmjF.23.0270, 4 other L major enzymes also had hits with the human genome, but that they were not annotated as CCDS by Ensembl. However, in these 4 cases (LmjF.18.1510, LmjF.18.1520, LmjF.26.2280, and LmjF.27.2440), homologies were also found with CCDSs at other genomic coordinates. In nr, we found homologous protein sequences for the human genomic hits corresponding to LmjF.23.0270, LmjF.18.1510, LmjF.18.1520, and LmjF.27.2440 that were not annotated as CCDS by Ensembl. We could retrieve a complete gene model only for EAX0113, which meant that the protein hCG2039601 between 19 998 852 and 19 999 097 bp on chromosome 18 escaped an Ensembl annotation. The gene models for the 3 homologies at human chromosomes 8 and 6 were partial, which indicates that they should likely be considered pseudogenes. Similarly, LmjF.26.2280 (EC 3.5.5.1—nitrilase) matched a pseudogene because an inframe stop codon (“*”) could be found in the sequence of the human homologous region (subject) in the tBLASTn alignment (Figure 4D).
Homo sapiens proteins retrieved from nr using the DNA stretch of the subject, which corresponded to the homologous region between the Leishmania major proteins and the human genome, as a query.

Abbreviation: NA, not assigned.
Chr. no. is for chromosome number.
Acc. no. is for accession number.
Pseudogenes.
Missing annotation.
The equivalent KEGG accession and ECs of the 17 human proteins from Ensembl that were homologous to the 14 L major enzymes (14 ECs and 3 repeats), given in Table 3, could be classified into 3 categories (Figure 4C). (1) Nine enzymes were annotated with 8 different ECs (1 repeat) in L major, which means that they were associated with different enzymatic reactions in L major and humans, ie, they could be considered specific from a functional standpoint. (2) Three L major enzymes were associated with 2 incomplete ECs in humans (1 repeat). (3) Five (1 repeat with the category “different EC numbers”) were annotated as enzymes in L major (4 ECs), but their human homologs were not annotated as enzymes (undef.) (see Table 3). It is worth noting here that in all the 17 cases, the homologous region between the L major protein and the human genome sequences precisely corresponded to a unique exon. These conserved regions, corresponding to the protein domains, were never interrupted by an intron in the human genome.
EC number annotations of the human proteins that are homologous to the Leishmania major enzymes classified as functionally specific.

Abbreviations: ATPase, adenosine triphosphatase; EC, Enzyme Commission; NA, not assigned; tRNA, transfer RNA.
KEGG accession numbers for L major.
KEGG accession numbers for Homo sapiens.
Ensembl accession numbers for H sapiens according to the format ENSG00000xxxxxx.
By comparing the α-helix and β-sheet distributions in the homologous pairs under “different EC number” and “incomplete EC number” (Table 3), we did not find any significant difference in the 2D profiles, which suggested that the classification of these sequences as functionally specific was ambiguous. It is only in the 5 sequences (4 ECs) under “EC number not assigned” (Table 3) that we found significant differences in the 2D profiles between both regions of the homologous pairs (Table 3, Figure 4F). However, among these 5 enzymes, only 2 (LmjF.14.0350 and LmjF.30.0180) were not involved in homologies with those of “different EC number.” These 2 enzymes in L major were composed of more β-sheets than α-helices, which did not occur in their human counterparts where the ratio between β-sheets and α-helices was approximately the same.
The relation of 2D distribution analysis between the L major and human conserved domains led us to finally consider 2 sequences from the 15 sequences (13 ECs) that were homologous to humans as being functionally specific enzymes in L major compared with H sapiens.
Considering the putative strictly specific enzymes of L major that shared ECs with the sequences classified as putative functionally specific enzymes, we found that 5 sequences (LmjF.18.0560, LmjF.19.1020, LmjF.26.0420, LmjF.36.1660, and LmjF.28.2100) shared 3 ECs (Table 4). Because these sequences are strictly specific, they are not expected to have human counterparts; however, the fact that they share ECs with the group of enzymes that are homologous to the human genome implies that they could be analogous to them. We also disregarded these 5 sequences, as we were only interested in filtering strictly specific enzymes in this report.
Putative functional and strictly specific enzymes of Leishmania major that share ECs.

Abbreviation: EC, Enzyme Commission; NA, not assigned.
ECs and Ensembl accession number for human sequences homologous with the putative functional specific enzymes of L major.
When a human EC was not available under “EC number not assigned,” we gave the one available under “different EC numbers” (Table 3) following the pattern “different EC numbers”/“EC number not assigned.”
We applied the same pattern as in “b” for Ensembl accession numbers.
When an Ensembl protein is homologous to a putative functional specific enzyme of L major is available, their pairing relationship must be about homology; when such an Ensembl protein is not available, there is no homology and the pairing is only about enzymatic function (EC), ie, analogy.
Strictly specific enzymes of L major
By further checking for remote homology with HHpred and HHsearch (Figure 4I), 7 sequences (LmjF.07.0270, LmjF.14.0180, LmjF.16.0530, LmjF.17.0140, LmjF.18.0200, LmjF.33.2540, and LmjF.36.3590) (6 ECs) were considered distant homologs and were filtered out from the putative strictly specific enzymes approved under Figure 4H (47 sequences, 29 ECs).
A homology analysis of the remaining 40 sequences (23 ECs) (Table 5) among Tritryps and Plasmodium falciparum shows that L major (query) shares the following: (1) 9 sequences with 24 sequences from T cruzi, (2) 7 sequences with 18 sequences from T brucei, and (3) 2 sequences with 1 sequence from P falciparum (Table S2).
Strictly specific enzymes of Leishmania major after hidden Markov model filtering.

Abbreviations: EC, Enzyme Commission; FAD, flavin adenine dinucleotide; UDP, uridine diphosphate.
EC numbers without any previous report in the Tritryps literature for drug development.
From the 40 strictly specific enzymes (23 ECs) (Table 5), 32 (19 ECs) were further disregarded because they did not seem to match any essential enzymes with a central position in the metabolic maps, as documented by KEGG (Figure 4J).
Only 8 specific enzymes, corresponding to 4 ECs (EC 2.1.1.41 with 2 paralogous sequences: LmjF.36.2380 and LmjF.36.2390; EC 2.7.9.1 with 1 sequence: LmjF.11.1000; EC 6.3.1.9 with 3 paralogous sequences: LmjF.23.0460, LmjF.27.1870, and LmjF.36.4300; and EC 6.5.1.3 with 2 paralogous sequences: LmjF.01.0590 and LmjF.20.1730), could be considered as potential target candidates for drug development. The complete relation of gene paralogy, analogy, and uniqueness of the 67 sequences released by AnEnPi as putative specific enzymes of L major compared with human is given in Table S3.
Evolutionary relationship of key strictly specific enzymes in L major with other human parasites
To better understand the consequences of the evolutionary relationship of key strictly specific enzymes for drug development (Figure 4K), we searched for homologous proteins in other human parasites (Table 6). It appeared that EC 2.1.1.41 (sterol 24-C-methyltransferase) is conserved among fungi and L major, which is expected from their common use of the ergosterol pathway. Interestingly, the homologies for EC 2.7.9.1 (pyruvate phosphate dikinase) are only shared among protozoa; the enzyme from Mycobacterium tuberculosis must be considered analogous because it does not share homologies with the L major sequences. As can be deduced from Table 6, EC 6.3.1.9 (trypanothione synthetase) is strictly specific to Tritryps, which confirms previous investigations. At first glance, EC 6.5.1.3 (mitochondrial RNA–editing ligase) is a function that appears relatively well conserved among human parasites. However, REL1 (LmjF.01.0590) and REL2 (LmjF.20.1730) are not homologous to the transfer RNA (tRNA)-splicing ligase (RtcB) of P falciparum and Toxoplasma gondii (Apicomplexa), even if they were annotated with the same enzymatic function (EC 6.5.1.3). Thus, the RELs from Trypanosomatidae and RtcBs from Apicomplexa should be considered analogous. The phylogenetic tree in Figure 5 shows that REL1 is conserved into Metakinetoplastina as far as B saltans and T borreli are representatives of this subclass, together with Trypanosomatidae. REL2 has not been taken into consideration here because it was proven to be nonessential in T brucei. 52
Distribution of strictly specific enzymes in human parasites.
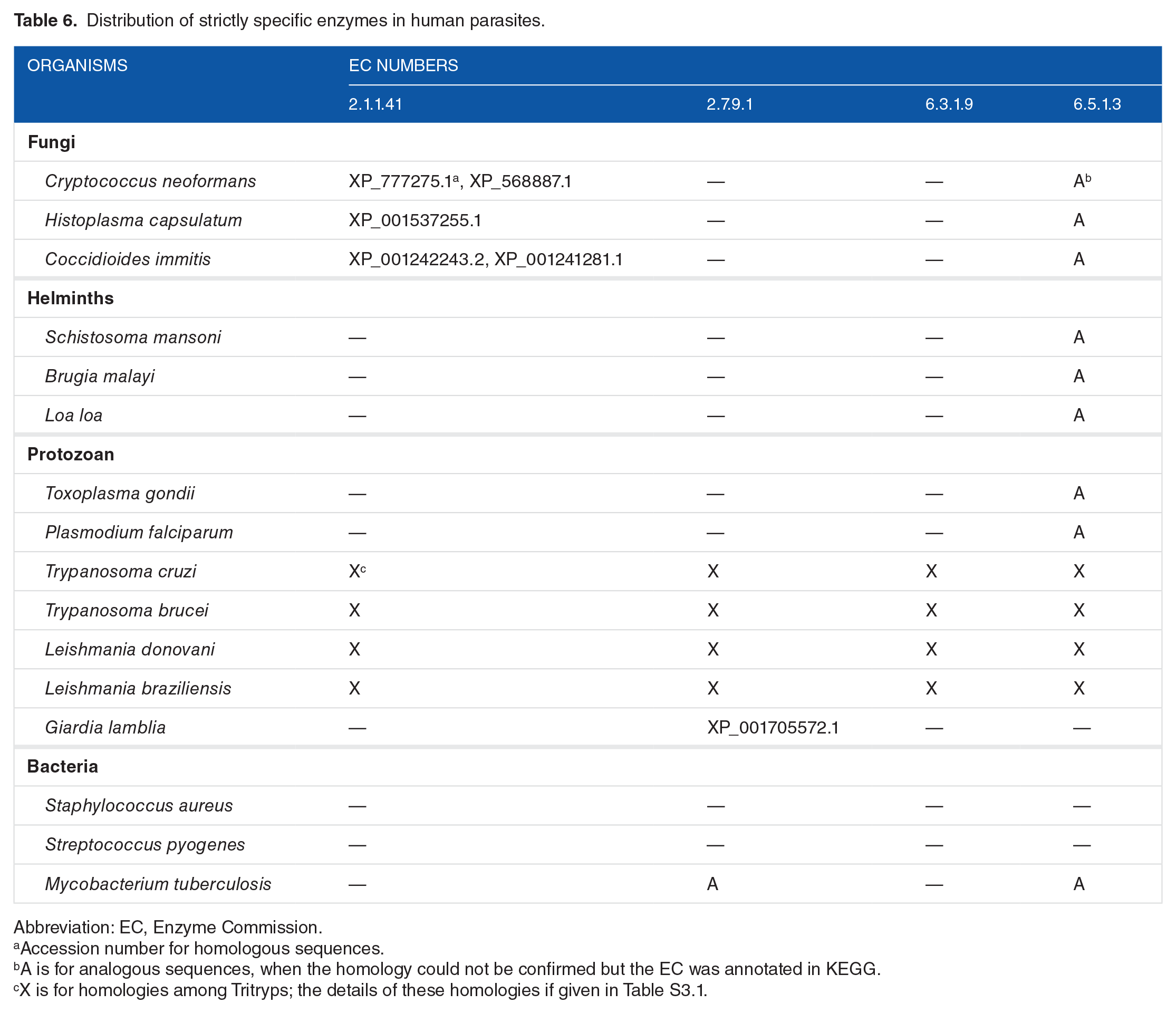
Abbreviation: EC, Enzyme Commission.
Accession number for homologous sequences.
A is for analogous sequences, when the homology could not be confirmed but the EC was annotated in KEGG.
X is for homologies among Tritryps; the details of these homologies if given in Table S3.1.

Phylogenetic tree of REL1 in Metakinetoplastina as obtained by maximum likelihood. Scale bar: 0.05 substitutions per site.
Discussion
Classification challenges and annotation ambiguity
In this study, we described a process for filtering out the false-positive annotations of enzymatic specificity in the host-parasite interactions released by AnEnPi. 32 The identification of specific protein targets in parasites (here, L major) compared with their host (here, H sapiens) is important for the development of drugs with the least amount of negative collateral effects for the host as possible. The procedure presented here is systematic, compatible with automation and suitable for host-parasite interactions that involve a lower eukaryote as the parasite and a higher eukaryote as the host. The fact that the targets identified here have been previously identified suggests that the results are valid and increases our confidence that the proposed methodology can be applied to other host-parasite systems with a lower level of associated knowledge.
We analyzed 67 protein sequences that correspond to 42 enzymatic activities and were previously classified as specific to L major compared with H sapiens by AnEnPi. A careful homology comparison of these sequences with those of Ensembl using BLAST allowed us to identify 15 significant homologous pairs between L major and H sapiens. These homologous pairs were associated with different ECs in L major and H sapiens, suggesting that they should be classified as functionally specific in L major compared with H sapiens. Of course, the classification of enzymes with functional specificity is completely dependent on the EC annotations, and it may result in trivial errors, as is the case for the ATPases that may carry H+ or Na+ cations through cell membranes. The 2 ATPases carrying H+ or Na+ are homologous, and the difference in their EC numbers is associated with the transported cations (the reaction and not the enzymatic activity), which are not associated with the ATPase activity itself when the substrate is adenosine triphosphate (ATP). Thus, care must be taken when interpreting ECs. In any event, we only found minute differences when considering the profiles of 2D structures between the sequence members of the BLAST homologous pairs.
Among the 15 putative functionally specific enzymes, the ECs of 9 L major proteins only differed from those of H sapiens in the fourth digit. When comparing the 2D alignments of these 9 homologous pairs, we only found slight differences, and it is difficult to claim that a drug for these L major targets would not have some kind of unwanted interaction with the human form that could result in negative side effects for the patients.
The Ensembl database involves processes of automatic gene annotation that are eventually manually curated; this is also the case for Havana.38,58 The AnEnPi pipeline is completely automatic, and it classifies enzymes as specific, homolog, or analog according to their homology and enzymatic activities. Obviously, incomplete enzymatic annotations lead to nonoptimized classifications by AnEnPi. The purpose of this study was to detect these cases and to provide more knowledge for improving the general performance of the automatic AnEnPi classifier.
Although sequencing methods have been producing large amounts of data, the genome assemblies based on these data may be incomplete or approximate. 59 Low-quality assemblies result in low-quality annotations60,61 and promote both over- and underestimations of the numbers of genes. 62 Considering the subject organism in a comparison, the incomplete assembly of its genome may lead to gene loss and thus generate a virtual specific gene in the query genome. It is possible to check the consistency of an event that is observed, but it is almost impossible to check the consistency of an event that is not observed. Thus, in this study, we did not take missed assemblies into account as a source of false positives. Only an in vitro analysis of our set of putative strictly specific enzymes can address this question.
For example, the existence of pseudogenes can be virtual or real, depending on whether they are the consequence of a mistake in the sequencing (induced frame shifts and premature stop codon) or assembling (gene cut) processes that lead to the inaccurate annotations.63–65 Indeed, many true pseudogenes have some form of biological activity, and thus their accurate annotation is potentially important to drug development. 65 We observed only 5 cases of a human pseudogene associated with the proteins of L major.
The homologies obtained by implementing HHpred and HHsearch have a higher confidence level compared with BLAST because HMMs consider the similarity in the amino acid sequence pairs from the L major query and the human sequence as well as the 2D and 3D structures of each sequence.
Enzyme essentiality
Enzymatic activity can play a key role in a metabolic pathway. Every pathway is associated with enzymes and their ECs. Therefore, a metabolic pathway is an oriented graph whose vertices are characterized by an enzyme and its associated chemical reaction. 66 A key enzyme is an enzyme that is contained in a path, lacks an alternative option, and plays an essential role in the survival of an organism. Consequently, the inhibition of a key enzyme necessarily results in the inhibition of the corresponding pathway and the organism’s debilitation. However, if a pathway that does not play an important role in the survival of the organism is targeted, one cannot inhibit the pathway, and an organism can continue to reproduce normally. Thus, the key feature for exploring a metabolic pathway is to identify the enzymes that play a key role in a pathway and to determine whether they are essential for the survival of the parasite, ie, whether their inhibition is deleterious to that organism. In the context of drug development, the ideal drug should be the one that can inhibit the activity of a specific enzyme in an essential pathway of the target organism, with no alternative route.
Functional specificity
Strict specificity, ie, the existence of a given enzyme in the parasite and not in the host, is obviously the best situation. However, functional specificity, where substrate specificity can be identified for the parasite form compared with the host form, can be contemplated as well. Actually, EC 1.3.1.71 (L major), which is associated with the steroid biosynthesis pathway, is an example of functionally specific enzyme that is homologous to the human enzyme EC 1.3.1.70. EC 1.3.1.71 could be explored for drug development as antifungal and antitrypanosomal agents. 67 Xu et al 68 showed that the knockdown of the gene encoding the 14-α-demethylase enzyme (EC 1.14.13.70), which is upstream of EC 1.3.1.71, in L major initially contributed to a dramatic change in the profile of the lipid composition in amastigotes and to virulence attenuation, which was reversed after a few weeks. Because the activity of EC 1.14.13.70 is common to the route of cholesterol and ergosterol syntheses in mammals and Leishmania spp, respectively, it is possible that L major got around the loss of EC 1.14.13.70 function in the knockdown parasites using sterols from the host. This escape strategy would be unlikely to occur if EC 1.3.1.71 was inhibited instead of the 14-α-demethylase enzyme activity in Leishmania spp, as EC 1.3.1.71 is inserted in a pathway of ergosterol synthesis that is specific to Tritryps. We did not find other potentially reliable functionally specific cases in this study because additional alternative metabolite routes may be available to the parasite to sustain the function that was inhibited by drug treatment. 68 More recently, McCall et al 69 demonstrated the importance of 14-α-demethylase enzyme in L donovani.
Strict specificity
Strictly specific enzymes can be divided in targets with an a priori lower (the 32 non–key enzymes) and a higher (the 8 key enzymes) potential for drug development.
Considering the strictly specific enzymes with a higher potential for drug development, our analysis showed that 4 enzyme functions, namely, sterol 24-C-methyltransferase (EC 2.1.1.41), trypanothione synthetase (EC 6.3.1.9), pyruvate phosphate dikinase (EC 2.7.9.1), and mitochondrial RNA–editing ligase (EC 6.5.1.3), can be considered for the development of a drug cocktail.
Unlike humans who have cholesterol in their biological membranes, trypanosomatids use ergosterol. Therefore, trypanosomatids present distinct enzymes in their sterol biosynthesis pathway. 70 Sterol 24-C-methyltransferase (EC 2.1.1.41) catalyzes a methylation of carbon 24, which is fundamental for ergosterol biosynthesis. The 24-C-methyltransferase reaction is inhibited by 22,26-azasterol, which causes morphological changes and lysis to Leishmania spp. 67
Trypanothione synthetase (EC 6.3.1.9) is fundamental to the synthesis of trypanothione, a redox metabolite of trypanosomatids that is involved in a number of processes, such as the regulation of intracellular thiol redox balance, drug resistance, defense against chemical agents, and oxidative stress. 71 The enzymes trypanothione synthetase and trypanothione reductase are involved in the trypanothione biosynthesis and metabolism pathways and are under study for the development of an alternative chemotherapy against trypanosomatids. 72
Pyruvate phosphate dikinase (EC 2.7.9.1) is an enzyme present in trypanosomatid glycosomes. 73 It was reported that this enzyme plays a key role in maintaining the balance of ATP vs adenosine diphosphate in the organelles in procyclic T brucei 74 and in the gluconeogenesis process in the amastigotes of L mexicana. 75 Therefore, it is believed that the pathway that includes pyruvate phosphate dikinase is central to the energetic metabolism of L major and that this enzyme deserves to be considered for leishmanicidal drugs.
The L-complex has been isolated from T brucei and Leishmania tarentolae mitochondria. At least 16 protein components have been identified, including REL1 and REL2. The precise function of REL1 and REL2, both annotated as EC 6.5.1.3, has been described by complementation of the knockout strains.76,77 In T brucei, the conditional disruption of REL1 is lethal in vivo as it affects both U-deletion and U-insertion editing resulting in an overall decrease in RNA size. 78 However, the loss of REL2 has no effect on viability or on editing. 79
These enzymes act in different metabolic pathways and cellular processes: sterol biosynthesis and membrane structure (EC 2.1.1.41), glutathione metabolism and oxidative stress response (EC 6.3.1.9), energy metabolism and carbon fixation pathways in prokaryote metabolism (EC 2.7.9.1), and RNA editing/posttranscriptional RNA processing (EC 6.5.1.3). Therefore, the simultaneous inhibition of these enzymes and consequently these pathways/cellular processes would likely promote an irreversible collapse of L major cells.
Using a strategy based on target druggability, Crowther et al 80 identified the enzymes LmjF.27.1870 (EC 6.3.1.9), LmjF.36.2380, and LmjF.36.2390 (the 2 latter having EC 2.1.1.41) as being potential targets for drug development in agreement with our results. However, these enzymes were later disregarded because they were not found to be orthologous to the enzymes classified as essential in other model organisms (Caenorhabditis elegans, Escherichia coli, M tuberculosis, and/or Saccharomyces cerevisiae) by bench experiments, perhaps these enzymes were not classified as essential because they did not exist in these organisms, as shown in Table 6 for M tuberculosis.
By contrast, based on an interactome inference, Flórez et al 81 identified 142 specific targets in the signaling network of L major compared with H sapiens. The only EC among our 8 strictly specific targets that we found in common with their list is specifically EC 2.1.1.41 (LmjF.36.2380 and LmjF.36.2390), which seems to contradict the statement by Crowther et al. 80 In addition, methyltransferase has been identified as a promising drug target in Cryptococcus neoformans. 82 Moreover, Goto et al83,84 described a vaccine candidate for the enzyme sterol 24-C-methyltransferase against visceral leishmaniasis that is effective against L infantum and L donovani.
Unlike Crowther et al, 80 we evaluated the essentiality of an enzyme by assessing its centrality in a metabolic pathway in the specific case of L major using its metabolic representation in KEGG.
Thus, the 8 strictly specific enzymes represented by the 4 ECs that we found to be essential are also reported as essential in the literature. By contrast, looking into Research and Training in Tropical Diseases (TDR; http://tdrtargets.org/), we found that 5 of the enzymes are considered essential for one or more trypanosomatids, 2 (LmjF.36.2380 and LmjF.36.2390) are not considered essential, and 1 (LmjF.36.4300) does not have data on whether it is essentially associated. 85
Gene redundancy and parasite resistance to drugs
We verified that, with the exception of EC 2.7.9.1, which has only 1 associated enzyme sequence, the other 3 EC numbers for strictly specific targets (EC 2.1.1.41, EC 6.5.1.3, and EC 6.3.1.9) have more than 1 gene representative in the L major genome. Gene redundancy for a same enzymatic function (see Table S2) is a potential source of drug resistance because a given inhibitor may inhibit a given enzyme, but not its putative isoforms, which could also be present. 86 Drug resistance may involve several different mechanisms, and a common solution to this problem is to increase the number of enzymes that are targeted by the inhibitors at the same time.
The process of gene accumulation, which has been called pyramidation, 87 is routinely used in classical breeding for plant resistance where it was first implemented.88,89 Pyramidation lowers the likelihood of virulence adaptation by a parasite because the corresponding accumulation of virulence genes becomes unsustainable in the given environmental conditions.
The idea of accumulating a number of targets that are simultaneously inhibited for a better disease control is a variation of the gene-for-gene relationship that has been described by Flor 90 and can be referred to as the gene-for-inhibitor concept. 91 Thus, formulating drugs into a cocktail should overcome parasite resistance; this practice is actually 92 already in use, as seen in the “Introduction” section. However, the formulation of a drug combination should also account for the dose-limiting negative side effects to normal cells and should protect the integrity of the host immune system. Thus, we sought to address this question here by identifying only the enzyme targets that are specific to L major, as targeting these candidates would most likely minimize the deleterious side effects from a therapeutic combination to patients. Of course, the results outlined in this report must still be confirmed by in vitro experimentation. Another question to be assessed in the future is the requirement for safety studies for each of the constituents of the combination treatment, independently and in combination, prior to the successful registration of that combination treatment; however, this question is beyond the scope of this report, which is mainly exploratory according to our current knowledge.
Drugs for key strictly specific L major enzymes
Drug reposition is a preferred route in drug development due to its savings in time and money. Unfortunately, an examination of TDR Targets did not show any approved drugs for the enzyme targets described here, but some inhibitors appear to be available at least for the experimental stages in Leishmania or other biological species. A search in the literature uncovered that (1) azasterols are inhibitors of sterol 24-C-methyltransferase (EC 2.1.1.41) in Leishmania67,92 and (2) azo dye naphthalene-like compounds, similar to suramin, are inhibitors of REL (EC 6.5.1.3).
93
Two of these compounds were later tested on the whole editosome and showed strong inhibition of U-deletion in RNA editing.
94
GW5074, mitoxantrone, NF 023, protoporphyrin IX, and
Under the hypothesis that the susceptibility of Leishmania would be stable in vitro to a combination of the compounds listed above, it would be possible to optimize their scaffold structure to reduce the overall cocktail toxicity to a human cell model. The engineering of this process is now facilitated by in silico modeling. A search for 3D models in PDB with sequences that are homologous to LmjF.36.2380, LmjF.36.2390, LmjF.11.1000, LmjF.23.0460, LmjF.27.1870, LmjF.36.4300, LmjF.01.0590, and LmjF.20.1730 showed that only the sequences of EC 2.7.9.1 and EC 6.3.1.9 were suitable for 3D modeling because their identity scores were larger than 39%, reaching up to a maximum of 97%. However, in the case of EC 6.5.1.3 and EC 2.1.1.41, the identity scores were too low (<25%) and the number of gaps were too high for successful 3D modeling.
Distribution of key strictly specific L major enzymes among human parasites
Sterol 24-C-methyltransferase (EC 2.1.1.41) is a component of the ergosterol pathway. This pathway has been extensively studied for drug development against fungi, and ergosterol pathway inactivation has been demonstrated as efficient and stable. The fact that sterol 24-C-methyltransferase can be shared with fungi means that drug reposition from fungi to Trypanosomatidae may occur, as was the case with fluconazole. Pyruvate phosphate dikinase (EC 2.7.9.1) is highly specific to Trypanosomatidae, and EC 2.7.9.1 was only found to be shared with M tuberculosis; however, the sequences did not align, which suggests that the enzymes of both lineages are analogous. The sequences of trypanothione synthetase (EC 6.3.1.9) of T cruzi showed identities between 37% and 58% with those of L major. These data agree with the literature, which demonstrates the expression of this enzyme and the presence of the corresponding trypanothione metabolism in T cruzi. 80 As expected, L major, which belongs to Tritryps, had a greater phylogenetic affinity with T cruzi and T brucei (Euglenozoa) than with P falciparum, which belongs to a different phylum (Apicomplexa). The status of sequence analogy that we gave to EC 6.5.1.3 (REL1) of P falciparum and T gondii is not surprising, considering that both species belong to the clade of Alveolates (Chromalveolates), whereas Trypanosomatidae belongs to Euglenozoa (Excavates), which are as far away from one another as they are from Animalia (Unikonts) in evolutionary terms (http://tolweb.org/Eukaryotes/3). However, the status of analogy only depends on the EC annotation. In that respect, one could ask whether an RNA-editing ligase that is involved in trans-splicing in Tritryps should have the same EC (EC 6.5.1.3) as a tRNA-splicing ligase that is involved in cis-splicing and tRNA repair, as the biological consequences of the 2 reactions are different in both cases. Interestingly, REL1 is conserved across the subclass of Metakinetoplastina, which shows its ancient emergence within Excavates. Unfortunately, we did not find a complete genome sequence for the Diplonema or Euglena from higher taxa to check the precise level of life history in which REL emerged. Apart from parasites, Metakinetoplastina also includes nonparasitic free-living forms, such as B saltans, which shows that REL function predates parasitic adaptation. Actually, REL is encoded by an informational gene and belongs to the primeval functions that were used by life as soon as the RNA world came into existence, 100 and it has been shown to have been invented at least 2 times independently.
Expression pattern of key strictly specific L major enzymes
Interestingly, the expression of LmjF.01.0590, LmjF.11.1000, LmjF.20.1730, LmjF.23.0460, LmjF.27.1870, LmjF.36.2380, LmjF.36.2390, and LmjF.36.4300 is described as being constitutive in the promastigote (blood) and amastigote (intracellular) forms of L major,101–103 which makes these sequences suitable targets for drugs that are effective against both parasite forms.
Conclusions
By applying a comparative method of enzyme function for the pathogenic relationship between L major and humans, we succeeded in restricting the number of specific enzyme targets that we believe are suitable for drug development. Of the 67 sequences (accounting for 42 enzymatic activities) that were classified as specific to L major compared with H sapiens by AnEnPi, only 40 (23 ECs) were strictly specific, as 27 enzymes (19 ECs) were disregarded for being ambiguous, functionally specific, or analogous. Among the list of 40 strictly specific enzymes, another 32 sequences (19 ECs) were also disregarded for not being essential in L major metabolism. Finally, we uncovered 4 enzymes (sterol 24-C-methyltransferase, RNA-editing ligase, pyruvate phosphate dikinase, and trypanothione synthetase) that are suitable for drug development with the purpose of minimizing treatment toxicity to the host. The method is systematic, includes a process for determining false positives, can be automated, and can be applied to the investigation of other host-parasite relationships.
Footnotes
Acknowledgements
The authors thank members of the Plataforma de Bioinformática Fiocruz RPT04-A/RJ for technological support and David William Provance Jr for reading and editing the manuscript.
Peer review:
Six peer reviewers contributed to the peer review report. Reviewers’ reports totaled 2767 words, excluding any confidential comments to the academic editor.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study is supported by fellowship from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (http://www.capes.gov.br/) to L.C.C., fellowship #11917-13-0 from Ciência sem Fronteiras (![]() ) to C.R.L., Grant Universal 14/2013 #480866/2013-9 from Ministério de Ciência e Tecnologia/Conselho Nacional de Desenvolvimento Científico e Tecnológico to A.C.R.G., and funding to N.C. under Instituto Nacional de Ciência e Tecnologia de Inovação em Doenças de Populações Negligenciadas #573642/2008-7.
) to C.R.L., Grant Universal 14/2013 #480866/2013-9 from Ministério de Ciência e Tecnologia/Conselho Nacional de Desenvolvimento Científico e Tecnológico to A.C.R.G., and funding to N.C. under Instituto Nacional de Ciência e Tecnologia de Inovação em Doenças de Populações Negligenciadas #573642/2008-7.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
LCC, NC, and ACRG conceived and designed the experiments; wrote the first draft of the manuscript. LCC, NC, CRL, MA-F, and ACRG analyzed the data. LCC and NC jointly developed the structure and arguments of the paper and wrote it with the contribution of MA-F. LCC, NC, CRL, ACRG, MA-F, PT, and PD agree with manuscript results and conclusions. LCC, NC, AFF, MA-F, PT, and PD made critical revisions and approved final version. All authors reviewed and approved the final manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.