
Abstract
Background:
Prostanoids are a family of lipid mediators formed from arachidonic acid by cyclooxygenase enzymes and serve as biomarkers of vascular function. Prostanoid production may be different in males and females indicating that different therapeutic approaches may be required during disease.
Objectives:
We examined sex-dependent differences in COX-related metabolites in genetically modified mice that produce a cyclooxygenase-2 (COX2) enzyme containing a tyrosine 385 to phenylalanine (Y385F) mutation. This mutation renders the COX2 enzyme unable to form a key intermediate radical required for complete arachidonic acid metabolism and provides a model of selective COX2 inhibition.
Design and Methods:
Mice heterozygous for the Y385F mutation in COX2 were mated to produce cohorts of wild-type, heterozygous, and COX2 mutant mice. We investigated whether the genotype distribution followed Mendelian genetics and studied whether sex-specific differences could be found in certain prostanoid levels measured in peritoneal macrophages and in urinary samples.
Results:
The inheritance of the COX2 mutation displayed a significant deviation with respect to Mendel’s laws of genetics, with a lower-than-expected progeny of weaned COX2 mutant pups. In macrophages, prostaglandin E2 (PGE2) production following lipopolysaccharide (LPS) and interferon gamma (IFNγ) stimulation was COX2-dependent in both males and females, and data indicated that crosstalk between the nitric oxide (NO) and COX2 pathways may be sex specific. We observed significant differences in urinary PGE2 production by male and female COX2 mutant mice, with the loss of COX2 activity in male mice decreasing their ability to produce urinary PGE2. Finally, female mice across all 3 genotypes produced similar levels of urinary thromboxane (measured as 11-dehydro TxB2) at significantly higher levels than males, indicating a sex-related difference that is likely COX1-derived.
Conclusions:
Our findings clearly demonstrate that sex-related differences in COX-derived metabolites can be observed, and that other pathways (such as the NO pathway) are affected.
Introduction
Prostanoids are a subclass of eicosanoids and include the prostaglandins, thromboxanes, and prostacyclins that are formed when the metabolism of arachidonic acid (AA) is initiated by 1 of 2 isozymes, commonly called cyclooxygenase (COX) enzymes, but officially known as prostaglandin-endoperoxide synthase (PTGS) or prostaglandin H2 synthase (PGHS) enzymes. COX1 is the constitutive form of the enzyme occurring in most cells under normal basal conditions, whereas COX2 is readily induced upon exposure to inflammatory stimuli such as cytokines, bacterial endotoxins, or hormones. The crystal structures of both COX1 and COX2 reveal that both COX enzymes contain a prosthetic FeIII-porphyrin in the peroxidase (POX) active site and a long hydrophobic channel, known as the cyclooxygenase (COX) binding channel, in which AA substrate binds.1,2 Both COX1 and COX2 are believed to occur as homodimeric proteins that are localized in the nuclear membrane and the luminal surface of the endoplasmic reticulum. 3 While structures and catalytic functions of COX1 and COX2 share similarities, they assume different physiological roles 4 with COX1 regulating vascular tone 5 and COX2 mediating inflammation, tumorigenesis, and atherosclerosis.6,7 The AA cascade is initiated by the actions of phospholipases leading to the release of AA from membrane phospholipids. AA, bound in the COX channel of either COX1 or COX2, is converted to a cyclic hydroperoxy endoperoxide, prostaglandin G2 (PGG2) and is reduced by the peroxidase (POX) active site to PGH2.3,8,9 As represented in Scheme 1, catalysis is likely initiated by oxidation of the FeIII-porphyrin by a peroxide and requires the involvement of a key tyrosine residue (Tyr 385) that is essential for COX activity.10,11 PGH2 is the precursor to a number of other biologically active molecules. Scheme 1 depicts the downstream metabolites that were the focus of investigation in this study that include PGE2 and thromboxane A2 (TxA2). PGE2 plays a role in many important processes, such as in the maintenance of the structure and function of the ductus arteriosus in fetuses and newborns, vasodilation, renal water absorption, fever, and pain.12-14 TxA2 is produced as a result of the action of platelet COX1 converting AA to PGH2 followed by catalysis of PGH2 to TxA2 by thromboxane A synthase. TxA2 is also produced by COX2 in inflammatory cells and by the kidney. TxA2 is regarded as a potent platelet aggregator and vasoconstrictor that is also involved in tumor-cell proliferation and invasion.15,16 TxA2 is an unstable compound and is quickly hydrated into TxB2 and converted by the liver into 2 metabolites: 2,3-dinor TxB2 and 11-dehydro-TxB2. All 3 metabolites (TxB2, 2,3-dinor TxB2, and 11-dehydro-TxB2) are excreted in the urine, but urinary 11-dehydro-TxB2 is a stable metabolite and can serve as an indirect measurement of the systemic biosynthesis of TxA2. Full reviews of the diverse families of oxygenated metabolites produced from arachidonic acid, including those that have not been explored here, such as other prostaglandins (PGs), prostacyclin (PGI2), leukotrienes (LTs), and lipoxins (LXs), can be found elsewhere.12,17
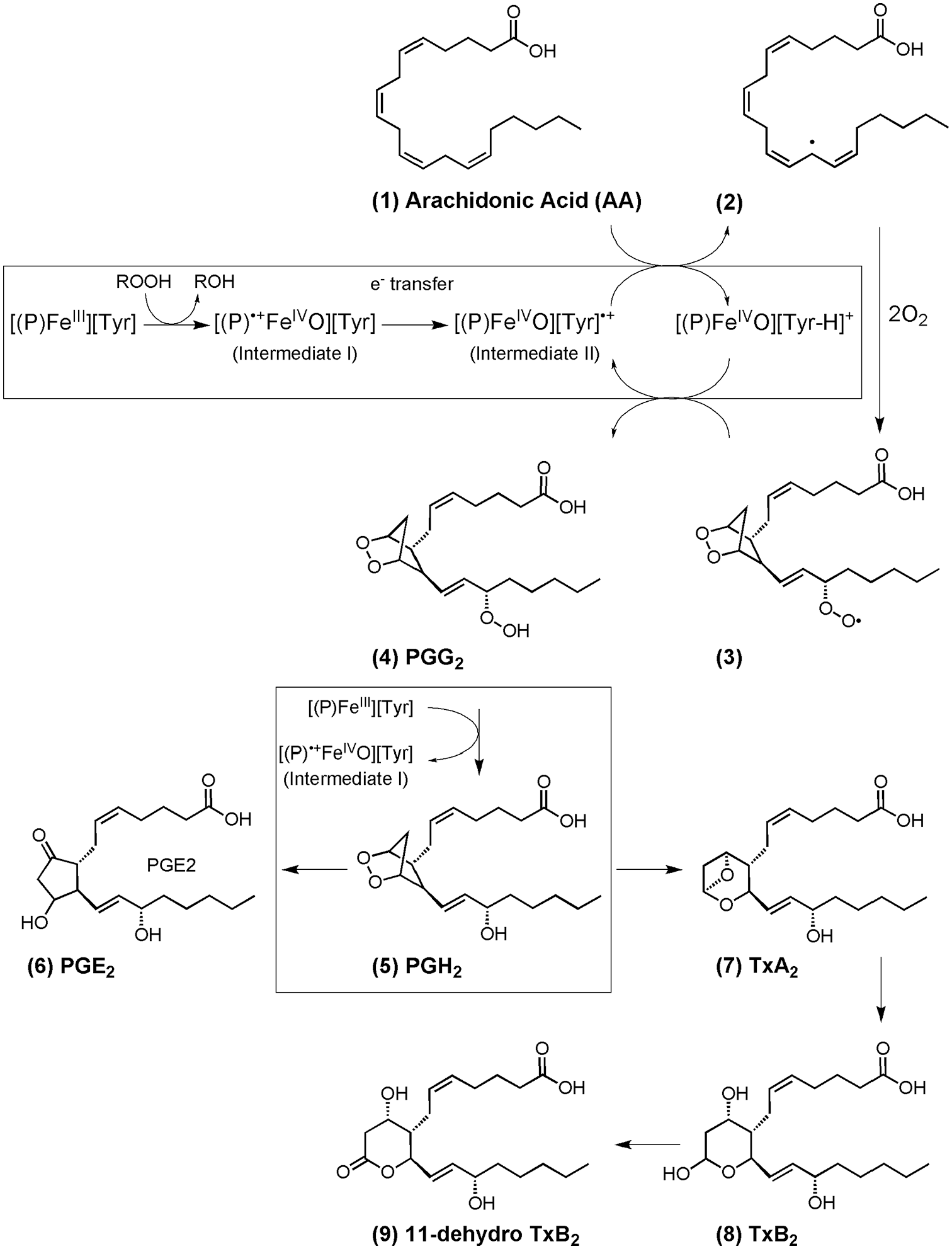
Catalysis is likely initiated by a peroxide (ROOH) leading to oxidation of the FeIII-porphyrin, denoted as [(P)FeIII], and resulting in the formation of an FeIV=O porphyrin π cation radical (intermediate I). Electron transfer occurs from a Tyr residue (Tyr385) located at the apex of the COX channel to quench intermediate I. When arachidonic acid (AA) (1) is bound in the cyclooxygenase channel of COX, the Tyr385 cation radical (intermediate II) abstracts the 13-pro(S)-hydrogen atom of AA forming a radical at the carbon 13 position of AA (2), and initiates oxygenation by O2 and cyclization to form a cyclic endoperoxide hydroperoxy radical (3) with subsequent prostaglandin G2 (PGG2) formation (4). This moiety is reduced by the peroxidase (POX) active site to PGH2 (5). Once formed, PGH2 can be metabolized in several different pathways including (but not limited to) one that involves PGE synthase and results in PGE2 formation (6), and another that utilizes thromboxane synthase and results in thromboxane A2 (TxA2) production (7). TxA2 is hydrolyzed in aqueous solution to TxB2 (8), and can be converted to the stable end product, 11-dehydro TxB2 (9). Boxed regions of the scheme indicate the POX-related activity of COX.
Inflammation is often treated using non-steroidal anti-inflammatory drugs (NSAIDs), most of which are non-selective COX inhibitors, such as aspirin, ibuprofen, indomethacin, and naproxen. However, long-term use of non-selective COX inhibitors in disease states such as rheumatoid arthritis and gout has been associated with gastric damage and peptic ulcer formation. 18 The development of COX2-specific inhibitors, such as Celecoxib (Celebrex) and Rofecoxib (Vioxx), were expected to relieve inflammation while sparing the ability of COX1 to produce prostaglandins that protect the gastric mucosa. Results from clinical trials evaluating COX2-specific inhibitors, however, attracted widespread attention due to an unanticipated incidence of myocardial infarction.19-21 As a result, Vioxx was withdrawn voluntarily from the world-wide market by Merck in 2004, and Celebrex now carries a “black-box” warning. 22 Studies have revealed that while COX2 is induced by pro-inflammatory stimuli, it is also constitutively expressed in tissues such as the brain, kidney, pancreas, intestine, and blood vessels where it may function to maintain homeostasis. 23
To investigate further the physiologic roles of the COX1 and COX2 isoforms, both COX1- and COX2-null mice were previously developed.24,25 Surprisingly, the absence of COX1 was not sufficient to cause spontaneous stomach ulceration indicating that the relationship between COX inhibition and ulceration is complex. 24 However, COX1 null mice exhibited a decrease in AA-induced platelet aggregation fitting well with the theory that since platelets express COX1 and not COX2, elimination of COX1 would result in reduced thromboxane A2 levels and thus, reduce the ability to induce blood clotting. On the other hand, the COX2-null mice were found to (i) possess a normal inflammatory response to treatments with tetradecanoyl phorbol or AA, (ii) show no innate gastrointestinal pathology, (iii) develop severe kidney disease, (iv) and be susceptible to peritonitis. 25 It was later shown that approximately 35% of the pups lacking COX2 died due a partially penetrant patent ductus arteriosus phenotype, although all the pups died if they were lacking both COX1 and COX2. 26
Thereafter, a mouse model that expresses a targeted knock-in mutation of COX2, in which tyrosine 385 was replaced by phenylalanine (Y385F), was developed.13,27 The Y385F mutation results in a COX2 enzyme that is still expressed but has no COX activity, although peroxidase (POX) activity is maintained. 11 The COX2 Y385F mutant mouse provides a model of selective COX2 inhibition, in that it mimics the effect of a COX2-selective drug that affects COX activity, without affecting the associated POX function. In this mouse model, it was demonstrated that lipopolysaccharide (LPS)-stimulation of peritoneal macrophages induced COX2 mutant protein to the same degree as native COX2 protein in wild-type (WT) macrophages, with no compensatory mechanism of COX1 expression. 13 Although COX2 mutant protein is expressed, COX activity is absent, but POX activity, as measured using N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD), remained intact and was no different to that observed in WT macrophages. A plausible explanation for the near normal neonatal survival of the COX2 Y385F mice compared to the COX2-null mice may be due to the formation of COX1-COX2 heterodimers in the ductus arteriosus, that is, an active catalytic subunit of COX1 combining with a mutant COX2 monomer that allows for remodeling at birth. 13 An alternative hypothesis to COX1-COX2 dimer formation is that the peroxidase component of COX2, which remains intact in mutant mice, also has an, as yet, unexplored role in ductal homeostasis. 13
Previously, we have shown that the FeCl3-induced injury model can be used to explore the hypothesis that male and female wild-type (WT) mice are not equally susceptible to thrombus formation. 28 Our results indicated that thrombosis formation in female WT mice was reduced compared to male WT mice and that this reduction could be correlated with increased induced NO synthase (iNOS) activity in female mice. During these studies, we also observed differential production of urinary prostanoid formation by male and female mice. 28
In this present study, we collected urine and elicited peritoneal macrophages that were stimulated with both LPS and interferon gamma (IFNγ) and studied whether sex-specific differences in the levels of prostanoids produced from wild-type and genetically modified COX2 Y385F mutant mice could be observed.
Materials and Methods
Mice
The breeding pairs of mice were originally obtained from Queen’s University, Canada. 13 The mice (from a mixed C57BL/6x129/Sv genetic background) contain a Y385F substitution in the enzyme cyclooxygenase 2 (COX2) that is expressed by the Ptgs2 gene. Mice heterozygous for the Y385F mutation in COX2 (COX2+/Y385F) were bred to obtain a cohort of WT (COX2+/+), COX2 heterozygous (COX2+/Y385F) and COX2 mutant (COX2Y385F/Y385F) mice. The mice were genotyped for the Ptgs2 allele by PCR analysis using kits (DNeasy® Blood & Tissue and Taq PCR Master Mix kits from Qiagen) and identified by tail tattoo. Primers that are complimentary to portions of exon-9 (EX9 Sense II TTG ACC AGA GCA GAG AGA TG, 20 bp), exon-10 (EX10 Antisense II CCA GAT TTG AGG AGA ACA GAT G, 22 bp), and neomycin (Neo Sense II GTT CCA CAT ACA CTT CAT TCT C, 22 bp) were used. The primers were mixed in equal parts in the Master Mix cocktail prior to PCR analysis of DNA extracted from murine tail tips. Wild-type (COX2+/+) and COX2 mutant (COX2Y385F/Y385F) mice gave rise to one band each, at ~566 and ~655 bp, respectively, whereas the COX2+/Y385F mice gave rise to both bands by gel electrophoresis. The mice were fed a regular chow diet on an ad libitum basis.
Inheritance of the COX2 mutation
The census data that recorded the gender and genotype of the pups from breeding male and female COX2+/Y385F mice over several years were analyzed to determine if the mutation introduced any deviation from Mendelian inheritance. The total numbers of male (193) and female (203) pups produced of each COX2+/+, COX2+/Y385F and COX2Y385F/Y385F genotype were determined and a chi-square (χ2) test was performed to assess if the observed data were compatible with the Mendelian model of inheritance. 29
Macrophage isolation, culture, and treatment
Macrophages were elicited from the intraperitoneal cavity following injection of sterile 4% thioglycollate medium (3 mL; Brewer Modified; Fluka) prepared in deionized water. After 4 to 5 days, the mice were euthanized under CO2 gas, and the abdominal skin was washed with 70% ethanol. Peeling back the skin exposed the outside of the peritoneum, after which ice-cold phosphate-buffered saline (PBS; 10 mL) was injected into the cavity. The peritoneal cavity fluid was retrieved using a fresh syringe barrel, and the solution was centrifuged (1500 rpm for 5 minutes at 4°C). The supernatant liquid was removed, and the cells were washed in PBS (5 mL) and centrifuged (1500 rpm for 5 minutes at 4°C). The washing procedure was repeated a second time, and following centrifugation, the cells were re-suspended in Dulbecco’s Modified Eagle Medium (DMEM; 2-6 mL) supplemented with 10% fetal calf serum (FCS), 1% glutamate, and 1% penicillin/streptomycin. The cells were counted using trypan blue and a hemacytometer, and then diluted appropriately with DMEM (containing supplements) to yield a cell population of approximately 5 × 106 cell/mL. The cells were dispensed into 6-well plates and allowed to adhere for 3 hours at 37°C with 5% CO2, after which the cell culture medium was replaced with fresh DMEM (containing supplements) and the cells were incubated at 37°C with 5% CO2 for 24 hours. In some cases, the cells were stimulated with lipopolysaccharide (LPS; from Escherichia coli 026:B6, 10 μg/mL; Sigma) and recombinant mouse interferon gamma (IFNγ; 100 U/mL; Calbiochem) to induce COX2 gene transcription. In other cases, cells were also treated with the non-specific NOS inhibitor L-NMMA (L-N-monomethyl-arginine; 250 μM). After 24 hours, the supernatants were collected and stored at −80°C for later analysis. The cells were washed with PBS (1 mL), lysed with 0.1 N NaOH (500 μL), and the protein concentration determined at a later date by performing a bicinchoninic acid (BCA1; Sigma-Aldrich) assay, according to manufacturer’s instructions. A total of 30 mice were used in this portion of the study (14 female and 16 male mice). The average age of the female and male mice employed was 27.4 ± 1.0 weeks. The average weights of the female and male mice were 21.2 ± 0.5 g and 27.1 ± 0.8 g, respectively.
Urine collection
Mice were placed in metabolic cages and urine was collected over a 24 hours period. The urine was sterile filtered (using syringes capped with a 0.22 μM Millex GP filter unit), aliquoted, stored at −80°C and later analyzed. In this study, urine samples from a total of 70 mice were collected, with the following numbers of mice in the different groups: 13 female and 12 male COX2+/+ mice, 12 female and 13 male COX2+/Y385F mice, and 12 female and 8 male COX2Y385F/Y385F mice). The average age of the mice was 34.5 ± 2.5 weeks.
Prostanoid and nitrite/nitrate measurement
Enzyme immunoassay kits (Cayman Chemical) were used to measure PGE2 and 11-dehydro TxB2. The 11-dehydro TxB2 kit measures both 11-dehydro TxB2 and 11-dehydro-2,3-dinor TxB2. The 11-dehydro TxB2 monoclonal assay buffer supplied with the kit is designed to convert 11-dehydro TxB2 into one conformation for more consistent results. Colorimetric assay kits (Cayman Chemical) were used to measure nitrate/nitrite and urinary creatinine. Urinary eicosanoid levels were normalized to creatinine.
Statistical analysis
Results are calculated as the mean ± the standard error of the mean (SEM). Significant differences are determined by t-test, with P < 0.05 defined as statistically significant. In addition, where applicable, the data were subjected to a one-way ANOVA, or a two-way ANOVA (with replication) analysis followed by a Tukey’s post-hoc assessment to determine whether differences between groups were statistically significant. The chi-square (χ2) test was performed using Excel. The analyses were performed using Prism GraphPad (version 4) and Excel (versions 14.7.7 and 16.16.27) software.
Results
Inheritance of the COX2 mutation
An analysis of the census data of weaned pups resulting from breeding male and female COX2+/Y385F mice over several years are provided in Table 1, and demonstrate a significant deviation from expected Mendelian genetics, indicating that the deviation must be due to some cause, and cannot be associated with chance alone.
Sex-specific inheritance of COX2 mutation.

A chi-square analysis of weaned female (203) and male (193) mice produced from breeding male and female COX2+/Y385F mice to determine if the mutation introduced any deviation from Mendelian inheritance.
The expected numbers of mice (*) are based on the Mendelian pattern of inheritance: 25% COX2+/+, 50% COX2+/Y385F, 25% COX2Y385F/Y385F.
It is evident that numbers of male COX2Y385F/Y385F pups are especially low compared to female pups, indicating some sex-specificity. In contrast, previous studies noted that COX2Y385F/Y385F mutant pups were obtained in the normal Mendelian ratio from heterozygous matings. 13 With complete COX2 gene disruption, COX2 KO mice were also born at the expected Mendelian ratio, although 57% of the neonatal pups died within 48 hours due to failure of closure of the ductus arteriosus after birth. 27 Notably, COX2Y385F/Y385F pups did not exhibit this problem, and it was suggested that the heterodimerization of COX1 and COX2 provided a compensatory mechanism to allow for normal closure that is not available to COX2-null mice. However, it was previously noted that the COX2Y385F/Y385F pups frequently died within weeks of birth due to kidney malfunction and peritonitis.25,27 In our study, the pups were not disturbed during the first week of birth, and while occasional deaths were noted (and it is possible that some pups were cannibalized), these fatalities did not occur to the extent that would account for the low numbers of COX2Y385F/Y385F pups. It is possible that the normal Mendelian ratio does result, but our observations may be skewed by some post-natal deaths and/or from prenatal lethality causing a significant reduction of COX2Y385F/Y385F pups, which has been observed in other mouse models. 30 Since the numbers of COX2Y385F/Y385F pups were low, it was necessary to conduct our research studies over several years in order to be able to study significant numbers of male and female mutant mice of the appropriate age. Further in-depth studies, however, are required to examine the extent of COX2 mutation involvement in prenatal fatality.
PGE2 produced by macrophages from female and male mice
Macrophages were elicited from male and female COX2+/+, COX2+/Y385F, and COX2Y385F/Y385F mice, and PGE2 production was measured in the supernatant media after 24 hours under 4 different conditions: (i) control, and in the presence of (ii) L-NMMA, (iii) LPS/IFNγ, and (iv) LPS/IFNγ + L-NMMA (Figure 1). Previous studies have shown that crosstalk between the prostaglandin and NO pathways occurs, and for this reason, we investigated the effect of NO on prostaglandin levels by using the non-specific NOS inhibitor, L-NMMA.31-33
PGE2 production by macrophages, elicited from male and female COX2+/+, COX2+/Y385F, and COX2Y385F/Y385F mice under control conditions (solid) and in the presence of L-NMMA (diagonal lines), LPS/IFNγ (dots), and LPS/IFNγ + L-NMMA (horizontal lines).
As expected, the biggest response in PGE2 levels was elicited upon LPS/INFγ stimulation and was dependent on the presence of COX2. The highest PGE2 levels, in response to LPS/IFNγ stimulation (in the presence and absence of L-NMMA), were produced by macrophages from mice in the following order: COX2+/+ > COX2+/Y385F > COX2Y385F/Y385F. In macrophages from COX2Y385F/Y385F mice that are completely deficient in COX2 activity, LPS/IFNγ treatment (in the presence and absence of L-NMMA) failed to induce PGE2 production above control levels. Thus, the response to LPS/INFγ stimulation is COX2-specific, as expected. For cells treated with LPS/IFNγ (in the presence and absence of L-NMMA), there were significant differences that showed a significant correlation with both sex and genotype, as measured by 2-way ANOVA. Tukey’s post hoc analysis indicated that a comparison of the values of any 2 different genotypes was statistically significant.
It has previously been found that NO production negatively regulates COX2 expression in response to inflammatory stimuli in smooth muscle cells and that iNOS gene deletion exaggerates COX2 induction. 31 Thus, inhibition of NO synthase was found to upregulate LPS/IFNγ–mediated COX2 protein expression, although specific sex-dependent effects were not investigated. 31 Our results indicated that the presence of L-NMMA significantly magnified LPS/IFNγ–mediated COX2 activity, but only in macrophages from female COX2+/+ and COX2+/Y385F mice. Conversely, in macrophages from male COX2+/+ mice, we noted a significant decrease in PGE2 production. These results indicate that the NO pathway does play a role in influencing PGE2 levels, and that the effect appears to be sex dependent. Within a particular genotype, there were small differences between male and female responses within the same treatment group that were statistically significant in some cases, as determined by t-test and indicated in Figure 1.
Basal levels of PGE2 production in control female mice across all 3 genotypes were similar and not significantly different. In contrast, basal PGE2 production in control male mice across all 3 genotypes showed small differences that were statistically significant. PGE2 production by macrophages from male COX2Y385F/Y385F control mice was significantly higher than from male COX2+/+ and male COX2+/Y385F mice. It is not clear whether the loss of complete function of COX2 in macrophages from male mice induces a compensatory mechanism whereby COX1 contributes to increased PGE2 activity. Such a determination was beyond the scope of the current investigation.
The results indicate that PGE2 produced above basal levels following LPS/IFNγ stimulation is COX2-derived, and that sex may also play a role in the levels observed. Since microsomal prostaglandin E synthase-1 (mPGES-1) plays a role in converting PGH2 to PGE2 in macrophages (Scheme 1), future studies to investigate the sex-dependence of this enzyme in macrophages would be useful.34,35 The results also demonstrate that inhibition of NOS activity by L-NMMA, may induce a sex-dependent response with regard to PGE2 production, although further investigation is warranted.
Nitrite/nitrate produced by macrophages from female and male mice
Nitrite and nitrate levels, produced by macrophages elicited from male and female COX2+/+, COX2+/Y385F, and COX2Y385F/Y385F mice, were measured in the supernatant media after 24 hours, under 4 different conditions: (i) control, and in the presence of (ii) L-NMMA, (iii) LPS/IFNγ, and (iv) LPS/IFNγ + L-NMMA.
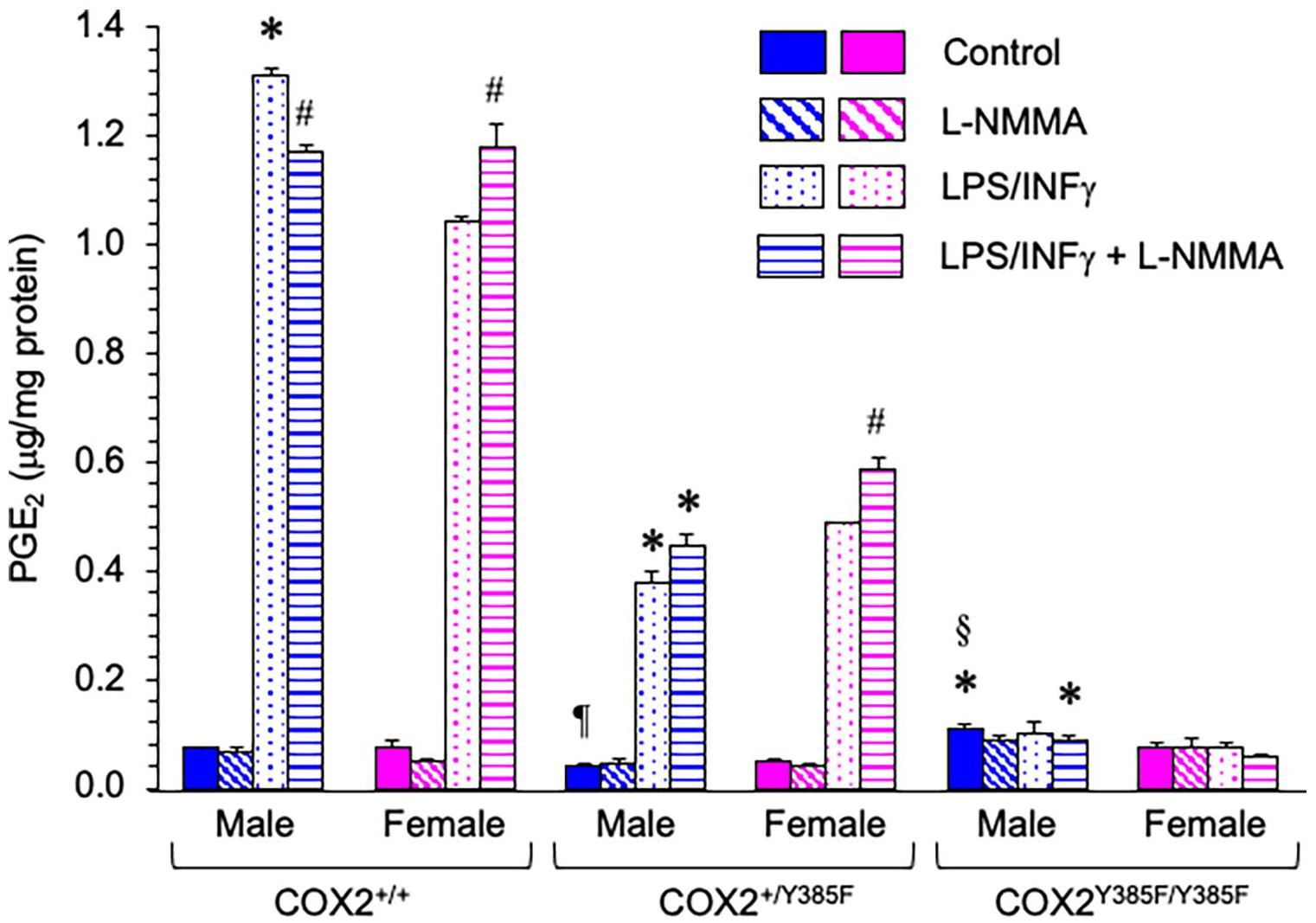
Treating macrophage cells with LPS/IFNγ increased nitrite/nitrate levels in a sex-and genotype-dependent way, as measured by 2-way ANOVA (Figure 2). Tukey’s post hoc analysis indicated that in response to LPS/IFNγ stimulation, there were significant differences between genotypes. Interestingly, macrophages derived from both male and female COX2Y385F/Y385F mice displayed a decreased ability to produce nitrite/nitrate following LPS/IFNγ stimulation, compared to COX2+/+ and COX2+/Y385F mice. By t-test, the biggest statistical difference in sex-related nitrite/nitrate production by macrophages in response to LPS/IFNγ stimulation was observed between male and female COX2+/+ mice, with males producing a greater amount. The effect of LPS/IFNγ stimulation was abrogated by the co-addition of the non-specific NOS inhibitor, L-NMMA.
NO production by macrophages, measured as total nitrite (NO2−) and nitrate (NO3−), elicited from male and female COX2+/+, COX2+/Y385F and COX2Y385F/Y385F mice under control conditions (solid) and in the presence of L-NMMA (diagonal lines), LPS/IFNγ (dots), and LPS/IFNγ + L-NMMA (horizontal lines).
As shown in Figure 2, there were no statistically significant differences between nitrite/nitrate amounts measured in control macrophages and macrophages treated with L-NMMA or LPS/IFNγ + L-NMMA from female and male mice across all 3 genotypes.
Overall, the results indicate that LPS/IFNγ-induced NO production is COX2-dependent, with a loss of COX2 leading to impaired LPS/IFNγ-induced NO production. Furthermore, LPS/IFNγ-induced nitrite/nitrate production was sex-dependent, with macrophages from male COX2+/+ mice displaying significantly higher levels than their female cohorts.
Urinary PGE2 produced by female and male mice
The COX2 Y385F mutant mouse provides a model of selective COX2 inhibition, and thus provides a way for us to examine the contribution of constitutive COX2 to urinary levels of prostanoids under normal physiological conditions. Urinary PGE2 levels were measured for male and female COX2+/+, COX2+/Y385F, and COX2Y385F/Y385F mice, and normalized to creatinine. As shown in Figure 3, slightly higher levels of urinary PGE2 were measured for female mice in all genotypes compared to male mice, but the sex-dependent differences only reached significance for urinary PGE2 obtained from male and female COX2Y385F/Y385F mice.

Urinary levels of PGE2 were measured for male and female COX2+/+ (solid bar), COX2+/Y385F (diagonal lines) and COX2Y385F/Y385F (dots) mice and normalized to creatinine.
A one-way ANOVA analysis for urinary PGE2 obtained from male mice across the 3 genotypes revealed that PGE2 levels are COX2-dependent, with a loss of COX2 activity resulting in a decreased ability to produce PGE2. Female urinary PGE2 levels remained indistinguishable across all 3 genotypes, and it thus, appears that this pathway under normal physiological conditions is COX2-independent for female mice. Further studies examining urinary PGE2 levels under conditions of inflammation, when COX2 expression is induced, are warranted.
Urinary 11-dehydro TxB2 produced by female and male mice
11-Dehydro-TxB2 is a proven reliable biomarker of in vivo synthesis of TxA2.36,37 Urinary 11-dehydro TxB2 levels were measured for male and female COX2+/+, COX2+/Y385F and COX2Y385F/Y385F mice, and normalized to creatinine. As shown in Figure 4, female mice across all the 3 genotypes measured consistently and significantly higher levels of urinary 11-dehydro TxB2 than the male cohorts. The COX2+/+, COX2+/Y385F and COX2Y385F/Y385F genotype did not significantly affect the levels of urinary 11-dehydro TxB2 measured for either male or female mice, indicating that COX2 does not affect the sex-dependent difference observed. Thus, the results suggest that the sex-related difference in urinary 11-dehydro TxB2 production is COX1-derived rather than being COX2-dependent. It would be interesting to explore whether urinary 11-dehydro TxB2 levels are altered under inflammatory conditions, when COX2 expression is induced.
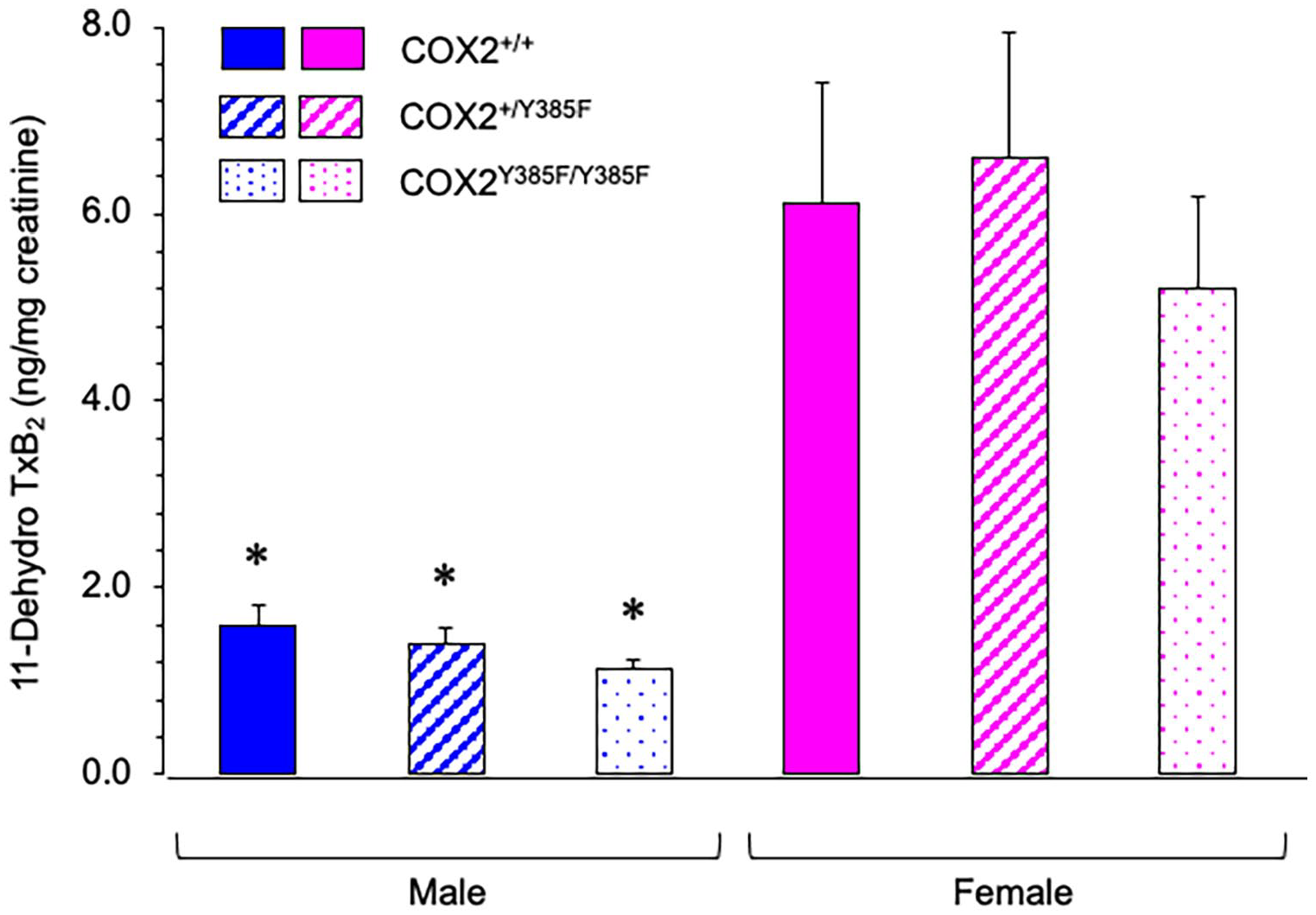
Urinary levels of 11-dehydro TxB2 were measured for male and female COX2+/+ (solid), COX2+/Y385F (diagonal lines) and COX2Y385F/Y385F (dots) mice and normalized to creatinine.
Discussion
Prostanoids are oxygenated metabolites of arachidonic acid that play important roles in a large variety of physiological functions that include being mediators of suppressors and activators of inflammation. Since inflammation is implicated in many disease states, the identification of altered levels of prostanoids can serve as biomarkers in the progression of certain pathogenic processes. 37 Examples of diseases that involve inflammation and have been correlated with changes in eicosanoid or prostanoid levels include atherosclerosis, 38 cancer, 39 psoriatic arthritis, 40 asthma, 41 chronic obstructive pulmonary disease (COPD), 42 and neurodegenerative disorders such as amyotrophic lateral sclerosis, Alzheimer’s and Parkinson’s diseases.43,44
Tracking eicosanoid and prostanoid levels and correlating their levels to specific disease states can provide targets for the development of improved therapeutic protocols, but efficient treatment can be made complicated by the fact that there might be sex-related differences associated with eicosanoid production. 45 Historically, sex and gender have been neglected in biochemical and pre-clinical research.46,47 For instance, while many chronic autoimmune diseases have been found to affect women to a greater extent than men, a survey of journal articles in this particular field from 2009 found that there was a strong bias toward the use of male animals.46,48 Furthermore, it has been noted that in studies examining the role of different cell types in the complex progression of certain disease states, such as atherosclerosis, many studies, if not all, do not report the sex of the cells employed. 49 Thus, research should not only consider the sex of the animals (or human subjects) but also whether isolated cells are derived from males or females. The sex-dependent differences in eicosanoid levels that have been observed in disease states indicate that males and females may require different therapeutic approaches.50,51 Furthermore, it is also important to consider that drugs may be metabolized differently by males and females with different sex-dependent side effects.46,52 A review of the sex differences in the pathophysiology and pharmacotherapy of inflammatory diseases related to eicosanoids has recently been reported. 45
Previous studies involving human neutrophils demonstrated that males produce higher levels of PGE2 than females, indicating a sex-dependence that has consequences for the inflammatory response. 53 In contrast, in a separate study, peritoneal macrophages elicited from adult female rats released, in a dose-dependent manner, significantly more PGE2 than macrophages from the male, upon stimulation, indicating gender differences in immune responses. 54 Herein, we demonstrate that LPS/IFNγ-stimulated macrophages from male and female mice produce different levels of PGE2 that are COX2-dependent (Figure 1). Small differences between males and females within similar treatment groups for a particular genotype also indicated that the response is sex dependent.
Inhibition of NOS with L-NMMA significantly enhanced PGE2 release in LPS/INFγ-stimulated macrophages (compared to LPS/INFγ-stimulated macrophages), but only in female COX2+/+ and COX2+/Y385F mice (Figure 1). It has previously been found that NO production negatively regulates COX2 expression in response to inflammatory stimuli in smooth muscle cells. 31 Thus, inhibition of NO synthase was found to enhance LPS/IFNγ–mediated COX2 activity, although specific sex-dependent effects were not investigated. 31 Furthermore, in our study, there was some indication that a loss of COX2 activity in male COX2Y385F/Y385F control mice led to the production of slightly higher PGE2 levels compared to COX2+/+ and COX2+/Y385F control mice, indicating that COX1 may be contributing to a greater extent in this case. In addition, it would be interesting to examine COX1, COX2, and NOS expression in macrophages obtained from male and female mice across all 3 genotypes in response to inflammatory stimuli.
It has previously been found that LPS increases both PGE2 and NO production in macrophages, and that the release of both can be attenuated by pre-treatment with the COX2 selective inhibitor, celecoxib. 55 In our study, we also demonstrated that NO production by LPS/INFγ-stimulated macrophages is COX2-dependent, as loss of COX2 impaired NO production by macrophages obtained from both male and female mice (Figure 2). While WT male macrophages produced a greater amount of NO than WT female macrophages upon LPS/INFγ-stimulation, this sex-dependence was not observed in macrophages from male and female mice lacking COX2 functionality.
Urinary PGE2 is mainly derived from the conversion of AA to PGH2 (Scheme 1) by either COX1 or COX2 in the kidney. While COX2 is traditionally regarded as an inducible enzyme, it is constitutively expressed at specific sites in the kidney. PGH2 to PGE2 conversion is catalyzed by different PGE synthases, of which cytosolic PGE synthase (cPGES) and microsomal PGE synthases type 1 (mPGES-1) occur in the kidney. 56 cPGES is believed to couple with COX1, whereas mPGES-1 is associated with COX2 and is upregulated together with COX2 in response to various pro-inflammatory stimuli.57,58 Prostanoid production in the kidney is important in the physiological control of vascular tone, renin release and blood pressure, and there is evidence that COX2-derived prostanoids regulate hemodynamics, as well as sodium and water reabsorption. 59 In contrast, elevated levels of COX2 and mPGE-1 in the kidney are associated with renal disease and cardiovascular risk, including hypertension and type 2 diabetes. 60 Thus a delicate balance of prostanoid production is required for healthy renal function, which is further made complicated by the existence of a sexual dimorphism in renal prostanoid production. 45 Deletion of mPGES-1 has been shown to reduce pain sensitivity,61,62 retard atherogenesis, 63 and fail to accelerate thrombogenesis, while suppressing PGE2 production and rediverting prostaglandin precursors to prostacyclin (PGI2) formation. 64 The results from such studies have prompted interest in developing drug targets of mPGES-1. 65
Our results (Figure 3) indicated that slightly higher levels of urinary PGE2 were measured for females, but values only became significantly different between male and female COX2Y385F/Y385F mice. While genotype did not appear to affect PGE2 production in female mice, there were significant differences observed for male mice, indicating a dependence on COX2. A loss of COX2 activity in male mice significantly decreased their ability to synthesize urinary PGE2. A similar trend in female animals producing more PGE2 than their male counterparts has also been observed in other studies.28,45,66,67 In spontaneously hypertensive rats (SHR), urinary excretion of PGE2 metabolites in female rats was higher than in males, indicating the presence of a sex-related difference in the renal prostanoid sytem. 66 Lower PGE2 expression in males was correlated with lower expression of both mPGES-1 and COX2 in the renal inner medulla and outer medulla, respectively. Further studies demonstrated that PGE2 production is testosterone-sensitive, rather than estrogen-regulated, as following orchiectomy, PGE2 metabolite excretion and mPGES-1 expression were both elevated in male rats. 66 Sullivan et al noted that since hypertensive males have a lower capacity than females to produce PGE2, selective COX2 inhibition would further cause a sex-dependent loss of PGE2 that could potentially cause severe kidney injury in males. 66 Our results (Figure 3) corroborate the finding that urinary PGE2 production in males is COX2-dependent.
Interestingly, it was previously demonstrated that a urinary PGE2 marker, known as tetranor-PGEM (11α-hydroxy-9,15-dioxo-2,3,4,5-tetranor-prostane-1,20-dioic acid) was depressed in both male and female COX2Y385F mice compared to wild-type mice but, in contrast to our results, significantly higher levels were produced by males compared to females. 64 Tetranor-PGEM production was also suppressed in mice missing mPGES-1 and in low-density lipoprotein receptor knockout mice, but its production was again markedly higher in males than in females.64,65 Furthermore, an earlier study conducted with human subjects also found that males produced higher levels of urinary tetranor-PGEM than females, although this difference was diminished in children and in an older (45-80 years) age group, implying that other factors may contribute to its availability. 68 Previous literature indicates that tetranor-PGEM is a major metabolite of both PGE1 and PGE2, with PGE1 arising from COX metabolism of dihomo-γ-linolenic acid, although the sex-dependence of its production is unclear.64,68 While the reason for the divergent results are not fully known, the different observations may be a consequence of analyzing a metabolite of both PGE2 and PGE1 (ie, tetranor-PGEM) in these previous studies64,65 rather than analyzing PGE2 (as measured by enzyme-linked immunosorbent assay) in our present study, which is widely used elsewhere for determining biological levels of PGE2.28,66,69 Furthermore, as remarked previously, our results concur with previous studies indicating that female animals produce more PGE2 than their male counterparts.28,45,66,67 The contrasting results, however, clearly demonstrate that further work is necessary, not just in delineating the factors that contribute to the sex-dependence of prostanoid production, but also in establishing the best ways to analyze these markers.
In other studies using mice that are prone to immune-mediated nephritis (DBA/1 mice), female mice were found to be superior in producing higher urinary prostanoid levels as compared to their male counterparts, and this increase was correlated with higher COX2 and mPGES-1 mRNA expression in the kidneys of female mice. 67 Female mice also showed a greater abundance in COX1 mRNA expression compared to male mice, but this trend did not reach statistical significance. mPGES-1 was found to be partially involved in urinary PGE2 production, since depletion of the mPges1 gene resulted in an approximate 50% reduction in urinary PGE2 in both sexes, indicating that other pathways also lead to PGE2 synthesis. These results further highlight that there are significant sex-related differences in prostanoid formation that would need to be carefully considered when offering therapeutic approaches to the treatment of inflammation and pain.
Herein, we also demonstrated that female mice produced higher levels of urinary 11-dehydro-TxB2 than males across all genotypes (Figure 4). Similar levels were observed for females across all genotypes and thus, did not appear to be COX2-dependent. While a sex-dependent difference in urinary 11-dehydro-TxB2 production is apparent, this effect is not driven by COX2, but is likely COX1-derived. This finding is in accord with previous results demonstrating that COX1 contributes significantly to urinary thromboxane levels (measured as 2,3-dinor-TXB2 metabolite) and that higher levels are found in female mice, indicating the presence of a sex-related difference in the renal prostanoid system.64,65 Urinary excretion of TxB2 has also been noted to be higher for female versus male spontaneously hypertensive rats (SHR). 66 However, systemic TxB2 did not reach statistical significance between males and females in SHR. 66 Interestingly, TxB2 excretion was not affected by gonadectomy in either male or female SHR, indicating that sex-related hormones do not contribute to its levels. 66 While higher levels of renal TxA2 might be expected to have a larger impact on blood pressure in females, it has been suggested that the concomitant increase in PGE2 in females may work to offset any impact of increased TxA2 on blood pressure, since female SHR have been shown to have a slower progression of renal injury compared with males. 66 However, the reason for and consequences of greater renal TxA2 production in female SHR remain unknown. 66 A recent study, however, found that sex differences play a role in the factors responsible for the regulation of vascular tone and contraction of coronary arteries in male and female pigs, and explain a role for TxA2 in females: TxA2 in perivascular adipose tissue was found to mediate contraction in females, whereas males experienced greater sensitivity to PGF2α. 70 It is not yet known to what extent these sex differences are maintained under pathological conditions, such as obesity, although obesity is known to enhance the contractile responses of perivascular adipose tissue.
Conclusions
Herein, we demonstrate that the COX2 Y385F mutant mouse provides a model of selective COX2 inhibition and can be used to examine sex-dependent differences that occur in the production of COX-related metabolites. Furthermore, this model can be used to examine the effects of COX2 inhibition on other pathways, such as the NO pathway. The findings reported herein clearly demonstrate that sex-related differences in COX-related metabolites can be observed, but further studies unraveling the mechanisms and the impact of these differences need to be performed. Our findings also highlight the need for future investigations examining prostanoid biology to consider sex as a variable not just in animal models, but also at the cellular level, such that it is possible to develop safe and efficient therapies in the medical treatment of both males and females.
Footnotes
Acknowledgements
RKU would like to thank Dr. Colin Funk (Queen’s University, Canada) for the donation of breeding pairs of mice heterozygous for the Y385F mutation in COX2 (COX2+/Y385F) that allowed colonies of mice to be established. In addition, RKU would like to thank Dr. Nigel Yarlett for help in maintaining the colonies of mice at Pace University. RKU is grateful for the financial support provided from Scholarly Research and the Provost’s Student-Faculty Undergraduate Research Awards from Pace University. KDJ, RSB and SS would like to thank Pace University for being the recipients of the Provost’s Student-Faculty Undergraduate Research Awards.