
Abstract
The standard of care in oncology has been genomic profiling of tumor tissue biopsies for the treatment and management of disease, which can prove to be quite challenging in terms of cost, invasiveness of procedure, and potential risk for the patient. As the number of available drugs in oncology continues to increase, so too does the demand for technologies and testing applications that can identify genomic alterations targetable by these new therapies. Liquid biopsies that use a blood draw from the diseased patient may offset the many disadvantages of the invasive procedure. However, as with any new technology or finding in the clinical field, the clinical utility of an analytical test such as that of the liquid biopsy has to be established. Here, we review the clinical testing space for liquid biopsy offerings and elucidate the technical and regulatory considerations to develop such an assay, using our recently validated PlasmaMonitorTM test.
Introduction
Over the last two decades, cancer diagnostics in the era of precision or personalized medicine has made tremendous headway with the recent advances in science and technology making it possible for early detection as well as non-invasive or minimally invasive monitoring, using liquid biopsies. Early detection and molecular characterization of tumors is crucial to a positive prognostic outcome by guiding therapeutic decisions. The current standard of care in genotyping solid tumor tissue for cancer patients to identify somatic gene variants for therapeutic action primarily begins with an invasive surgical biopsy procedure, limiting the longitudinal follow-up. In contrast, liquid biopsies being minimally invasive allow for evaluation of the patient at multiple time points during and post-therapy to assess treatment response as well as for the detection of recurrence, resistance, and minimal residual disease, in theory enabling timely interventions as deemed necessary for effective management of disease. They also offset the burden of a surgical biopsy and are extremely valuable in cases where a patient is diagnosed with an inoperable/inaccessible tumor or primary biopsy of tissue is not possible due to high risk of bleeding or nerve injury. 1
Depending on the cancer type and stage, a number of specimens such as blood (plasma, serum), urine, saliva, pleural effusions, amniotic fluid, nasal secretions, bronchoalveolar lavages, lymphatic and peritoneal fluids, bone marrow aspirates, cerebrospinal fluid (CSF), prostatic fluid, peritoneal lavage, sputum, gastric juice, breast milk, ascites and biliary, and even stool samples (Figure 1A) have been evaluated for their potential to serve as non-invasive surrogates of tumor monitoring, 2 with plasma being preferred across all cancer types. 3 CSF is preferred for tracking central nervous system (CNS) tumors 4 ; saliva for head and neck cancers 5 ; urine for renal cell carcinoma, bladder, and prostate cancer 2 ; and stool being used for colorectal and pancreatic cancer. 6 The main analytes being evaluated in liquid biopsies are extracellular vesicles (such as exosomes), circulating tumor cells (CTCs), and/or residual fragmented DNA 3 (Figure 1B).

(A) Specimen types and (B) analytes for liquid biopsy.
Exosomes are small round vesicles of endosomal origin, approximately 50-100 nm in diameter, carrying DNA, RNA, miRNAs, and proteins released by multiple cell types (including tumor cells) into the extracellular environment. 7 As genetic alterations promote the interphase of cancer cells, the cancer cells shed from the tumor as CTCs. As cells including CTCs undergo apoptosis, necrosis, and perhaps secretion,8–10 they shed fragmented DNA into the circulation. DNA released irrespective of cell of origin is called cell-free DNA (cfDNA), and when released specifically by cancer cells, it is referred to as circulating tumor DNA (ctDNA). Due to concerns of sensitivity and specificity, false negatives in particular, evaluation of CTCs is therefore not yet fully accepted into clinical practice for guiding treatment decisions.
cfDNA is a mixture of normal DNA, wild-type DNA from tumor, as well as mutated DNA from tumor, requiring high-sensitivity technologies for detection, particularly since circulating nucleic acids have variable circulating half-lives ranging from 15 minutes to several hours,11,12 with concentrations of 0-1000 ng/mL of blood in cancer patients and 0-100 ng/mL of blood in healthy individuals.13,14 Analyzing cfDNA might not provide information of the entire landscape of the tumor, especially keeping in mind tumor heterogeneity; however, if appropriate technologies are used, it does enable earlier detection with prognostic potential to a certain extent.15,16 Studies have demonstrated that serum has a 3-24 fold increase in cfDNA compared with plasma, as a result of hemolysis during clotting or from leukocyte contamination results, with the amount of contamination increasing with the number of days between blood withdrawal and serum separation.14,17 Increased cfDNA in serum is associated with a higher false positive rate, making plasma the preferred specimen for evaluation of cfDNA.
Evaluating the clinical utility of non-invasive techniques to detect molecular alterations in CTCs and cfDNA has been a topic of interest in precision oncology as it provides the potential to safely and cost-effectively monitor the tumor profile at multiple time points throughout the patient’s clinical course. This concept of longitudinal testing is made possible through the economics and practicality of easily accessible specimens such as blood, saliva, and urine. Furthermore, variable but positive evidence continues to emerge that demonstrates the potential benefit (and limitations) of capturing multiple observations of a tumor’s genomic profile over time. 18 High-frequency monitoring may also prove critical to a physician’s ability to tailor or change a therapeutic course based on the molecular evolution and diverse landscape of the tumor 18 ; however, the ability of liquid biopsy tests to predict response to therapy is still the subject of active investigation in clinical and preclinical research. When tumors progress and metastasize, tumor sites may become inoperable. Obtaining a tissue sample that provides a landscape view of the tumor can become highly unlikely without the use of a liquid biopsy-based assay. Glioblastomas and spinal cancers can progress quickly, and obtaining a small tissue sample via a surgical biopsy may not provide a quick enough turnaround time at a chance to treat the cancer. Even a moderate amount of hemorrhage that occurs in the biopsy tract could be potentially threatening to the patient’s neurological function or even life. 19 By implementing liquid biopsy technologies, a simple blood draw can provide real-time genetic and molecular information about the tumor without the invasive surgical procedure which provides tremendous benefit to inoperable cancers such as brain and spinal cancers. 20
The Current Landscape of Liquid Biopsy Testing
With personalized medicine increasingly expanding into the clinic, more therapeutics are actively being developed and used in patients based on the molecular profiles of their different cancer types which is why discovery and classification of these biomarkers is of utmost importance. Molecular evaluation of cfDNA in cancer patients has emerged as an attractive option offering multiple benefits in patients with diverse tumor types, with a highly precise evaluation of the tumor genomic alteration landscape, reflective of disease burden.21–23
Clinical assays are developed to address an unmet need of the patient, are geared to a specific disease area, and use standard or novel technologies available in a specific lab. Detection of cfDNA requires highly sensitive techniques due to the small fraction of tumor-specific DNA mixed with wild-type cfDNA and some amount of genomic DNA from white blood cells. Standard methods for the detection of known hotspot mutations are polymerase chain reaction (PCR) based and include nested real-time PCR, mutant allele-specific PCR, and digital PCR which include droplet digital PCR (ddPCR), BEAMing, and microfluidic digital PCR. 12 Targeted deep sequencing methods such as SafeSeq and CAPP-Seq are highly sensitive methods that allow for the detection of selected single nucleotide variants (SNVs), copy number variants (CNVs), and rearrangements across specific regions. 12 Next-generation sequencing (NGS) technologies are currently being applied to cfDNA analysis, which enables high-throughput, low-cost sequencing to identify alterations genome wide, including estimation of tumor mutation burden (TMB) and microsatellite instability (MSI).
Strategies for the evaluation of cfDNA depend on the end goal of identifying resistance, monitoring for minimal residual disease or prima facie looking for recurrence, to be able to implement appropriate treatment regimens or companion diagnostics as available. Toward that end, there are several tests currently available for clinical testing, ranging from hotspots in a single gene, full gene coverage, panels for specific cancers, or a combination of all the above (Table 1). The decidedly selective approach, often designed for the purpose of assessing highly recurrent loci for mutations that directly inform treatment decisions through on-label use of Food and Drug Administration (FDA)-approved drugs or recommendations based on clinical practice guidelines, involves evaluation of hotspots or single genes. An example being the Cobas® EGFR Mutation Test v2 which is FDA approved for use in non-small cell lung cancer (NSCLC) patients. Focusing on evaluation of 42 mutations in exons 18, 19, 20, and 21 in the EGFR gene, this test aims to help patients with NSCLC that may not be suitable to undergo an invasive procedure and a genetic test focusing on the EGFR gene alone may be beneficial for therapy. An expanded approach for patients with an unknown primary tumor site would involve using a panel of genes such as the Guardant360 assay, 24 which includes 73 genes in which all exons are completely sequenced to exploit the detection of known somatic variants. Similar to other tests currently available for clinical use that span the spectrum of hotspots, through single gene and/or encompass multiple genes (Table 1), this panel is used across different cancer types and specimens (blood and/or formalin fixed paraffin embedded-FFPE tissue) and can be used to identify multiple therapeutic targets.
Liquid biopsy assays currently available for clinical use.
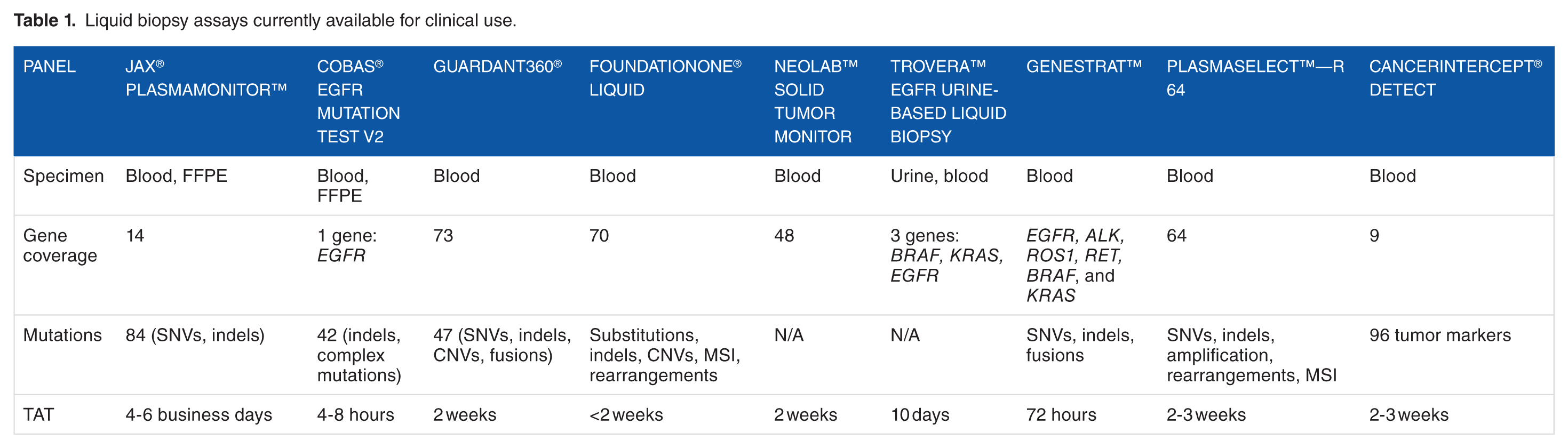
Depending on the approach taken, there are associated advantages, limitations, and challenges. In terms of clinical utility, single gene evaluation offers disease-specific coverage, a shorter turnaround time, is cost effective, and most importantly from a patient perspective, in the case of NSCLC as exemplified, is that the variants evaluated if detected are associated with targeted therapies. However, there are also limitations with single gene assays, in terms of the potential to miss variants in other genes not being evaluated. The FDA-approved Cobas® EGFR Mutation Test v2 panel as outlined above targets specific EGFR mutations in NSCLC patients exclusively, missing other mutations in genes such as ALK, BRAF, KRAS, MEK, and MET which also have potential to provide therapeutic yield. 25 Another concern with such an approach is the observation that the frequency of EGFR gene mutations in NSCLC patients when undergoing chemotherapy decreases 26 and therefore although approved by the FDA, this assay may not represent a suitable test for monitoring NSCLC patients receiving chemotherapy.11,26 When designing a single gene panel, identifying gene mutations with associated therapies is crucial but taking into consideration other factors that will impact a false-negative result is equally critical. While single gene panels offer a comprehensive view of the mutations that exist within that gene, significant data and information on the tumor can be lost without looking at possible mutations that may exist elsewhere and may be important for the treatment of the tumor.
Larger panels may not always be the best option in a clinical setting, as the cost increases and sequencing depth decreases per gene, the overall benefit to the patient can be uncertain but the greater potential to assess prognosis, detect recurrence, and get an insight into difficult-to-diagnose tumors remains present. 11 With a larger scope of genes to look at comes many variables and with that a multitude of challenges. In a recent cross-assay comparison, both the Guardant360 panel (Guardant Health Inc) and the PlasmaSELECT-R64 panel (Personal Genome Diagnostics) were run on identical metastatic prostate cancer patient samples to determine the reliability and potential utility of these assays. 27 To determine concordance across both tests, only genetic alterations in genes supposedly covered by both platforms (42 genes) were considered. Patients with one or more alterations demonstrated by at least one platform (31 of 40), whose all cfDNA alterations were covered by only one of the tests, were excluded (6 of 40). Only 3 (7.5%) of the 40 patients had complete congruence with one or more alterations. There are a number of reasons for the lower concordance including small sample size, variation in genes evaluated (regions of coverage, depth of coverage, etc), timing of different samples collected, but most importantly differences in limit of detection (LOD). This study suggests that the lack of concordance in genetic profiling across platforms and technologies could jeopardize the clinical benefit of personalized medicine. It also emphasizes that standardization of technologies and procedures across laboratories is needed to ensure consistency of results.
Development and Validation of the JAX PlasmaMonitor™
To provide comprehensive monitoring of cancers, the Jackson Laboratory (JAX) designed a non-invasive liquid biopsy assay to complement the current oncology test menu in the clinical laboratory. The JAX PlasmaMonitorTM was developed to cater to our patient pool which primarily consists of stage IV metastatic cancers and was therefore developed as a pan cancer panel (Figure 2). Being able to use the PlasmaMonitorTM to track the progression of cancer, monitor any arising clones that may develop resistance to treatment therapy, as well as recognizing new possible therapies as the tumor evolves is an undoubtable benefit. While maintaining the highest of standards, the assay was to include the most clinically relevant genes across a wide scope of cancer types (Figure 2A-C) that could be provided to the clinician in a reasonable amount of time, in a cost effective way, and was therefore designed to be a 14 gene, 84 variant hotspot pan cancer panel (Figure 2A).

PlasmaMonitor™ panel design, content, and criteria for inclusion. (A) Lists the 14 genes on the panel with specific hotspots across each gene; (B) outlines the coverage of the different variants across various cancer types; (C) represents the percentage coverage of panel across certain tumor types, using the number of samples mutated with those variants on our panel in a specific gene for that tumor type; (D) schematic for the logic that went into picking gene content; and (E) lists the criteria considered to determine actionability of a gene/variant.
To determine the coverage of the panel per cancer type, as an estimate of clinical utility, as the panel was designed, we used all the variants in catalog of somatic mutations in cancer (COSMIC) for each individual cancer type (such as breast cancer) to filter that list to contain only the variants present in the PlasmaMonitorTM for that tumor type. Data for the number of variants present in the panel identified for a specific gene in that tumor type and the number of samples mutated with those variants on our panel in a specific gene for that tumor type were narrowed down (Figure 2C). The goal was to provide an assay that could offset the disadvantages of a single gene and multi-gene panel while maintaining the benefits and integrity of what liquid biopsy panels are able to offer.
Panel design
To identify the most appropriate gene content for the panel, we scanned the literature to identify gene/variants with therapeutic, prognostic, and/or diagnostic impact; evaluated the positive yield rate from our solid tumor NGS panels; and collated the information to design a panel primarily based on the actionability of variants (Figure 2D). Actionability being defined as evidence of response or resistance to FDA approved therapies and/or investigational therapies, availability clinical trials recruiting for a specific gene/variant, and evidence of prognostic significance (Figure 2E). While the PlasmaMonitorTM is a smaller panel compared with some currently offered on the market (Table 1), it includes clinically relevant hotspot mutations in genes across multiple cancer types (Figure 2A-C).
Technical considerations
As elucidated so far, while genomic profiling for tumor mutations has been successfully demonstrated from cfDNA, the low amount of cfDNA in the blood, which can be less than 1% allelic frequency, presents significant challenges for reliable variant detection. As our NGS panel was developed, technical and analytical factors were evaluated to develop a test with high sensitivity and specificity, keeping in mind the LOD for low (<1%) variant allelic frequency (VAF).
Optimization of pre-analytical parameters such as plasma separation and selection of an isolation method that ensures extraction of a sufficient amount of high-quality DNA is critical. It has been shown that these pre-analytical factors of blood sampling and processing can strongly affect DNA yield 24 as well as downstream analysis. The development of PlasmaMonitorTM included evaluation of plasma separation and cfDNA extraction methods to maximize yield of quality DNA in a scalable, cost-effective manner. Introduction of an additional centrifugation step at 16 000 g for 10 minutes, post separation of plasma from whole blood resulted in decreased debris. We evaluated four commercial cfDNA extraction kits across 35 samples; QIAamp circulating nucleic acid kit (Qiagen), NucleoSpin Plasma XS (Macherey-Nagel), ZR Serum DNA kit (ZYMO Research), and the NEXTPrep-Mag cfDNA isolation kit (Bioo Scientific) (Figure 3A). Based on the yield, scalability, turnaround time for processing, and cost effectiveness, we opted for the NEXTPrep-Mag cfDNA isolation kit.
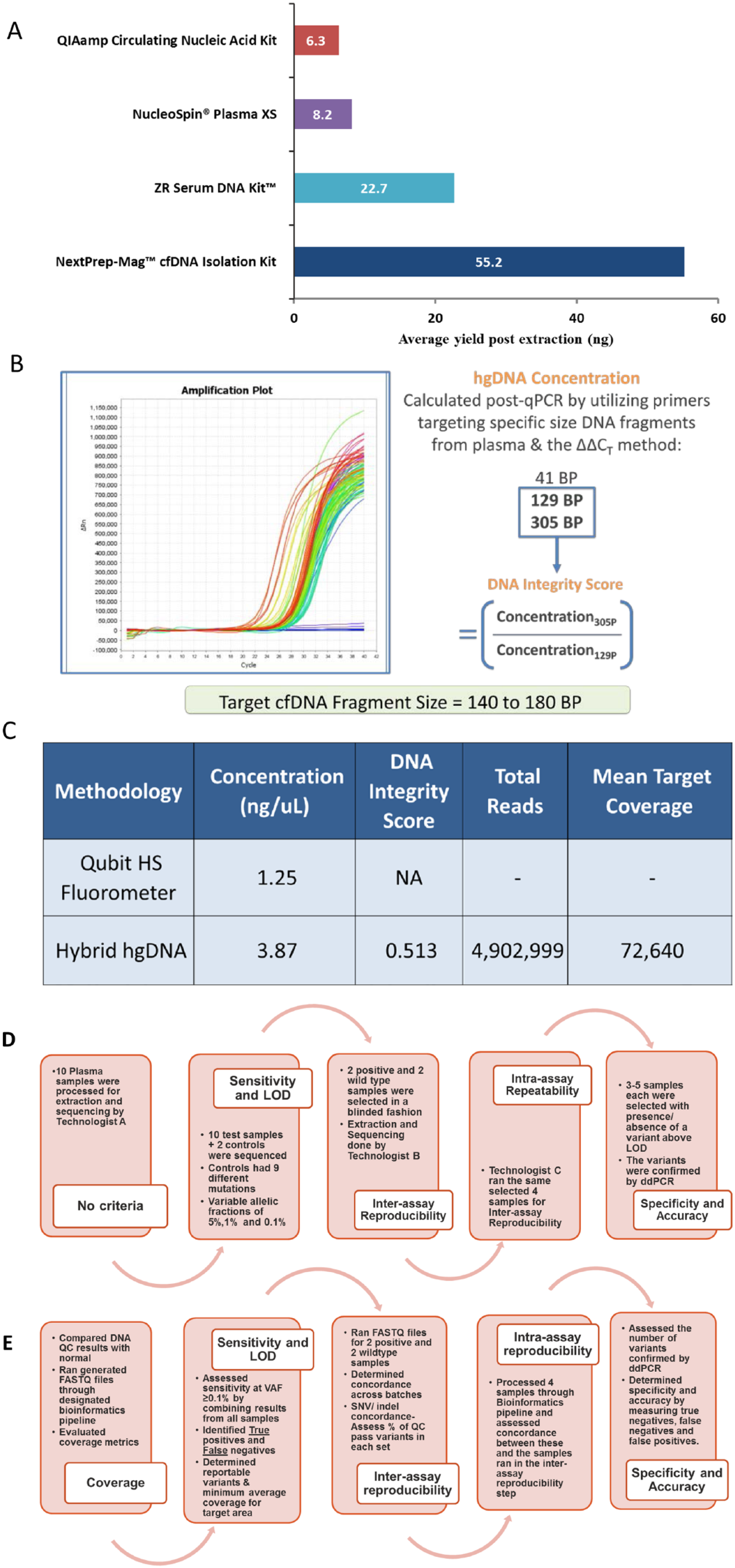
PlasmaMonitor™ pre-analytical and technical considerations. (A) Comparison of four commercial cfDNA extraction kits for yield; (B) quantification and qualification of the extracted cfDNA using a custom Human Genomic DNA QC Assay; (C) comparison data demonstrating that using our custom quality approach provides more robust quantification data in comparison with the standard Qubit assay; (D) pre-analytical validation steps including sensitivity and limit of detection (LOD), inter-assay reproducibility, intra-assay reproducibility, and specificity and accuracy; (E) post-sequencing validation steps including sensitivity and LOD, inter-assay reproducibility, intra-assay reproducibility, and specificity and accuracy.
Current cfDNA extraction methods yield modest quantities of nucleic acid content, emphasizing the importance of sensitive and accurate quantification and qualification of cfDNA in the molecular sequencing process. Although we realize that a bioanalyzer analysis to assess fragment size and quality is widely accepted to be the norm, we evaluated and validated the use of a modified Human Genomic DNA (HGD) QC Assay for quantification and qualification of the extracted cfDNA to enable effective downstream processing, taking into consideration DNA integrity index (DIN). To be able to establish this method as a QC step in our process, we performed a comparative evaluation of a Qubit Fluorometric 2.0 Assay vs a HGD QC Assay (Figure 3B). Cell-free DIN is measured as the ratio of long-to-short DNA fragments. 28 cfDNA from apoptotic cells is uniformly truncated into 180-200 bp fragments, whereas cfDNA from necrotic tumor cells varies in length, leading to elevation of larger fragment DNA. 29 Our results clearly demonstrated that using our custom HGD quality control approach provides more robust quantification of cfDNA in comparison with the Qubit assay. The method is also capable of generating a quality metric absent from the Qubit approach to assess for the presence of high molecular weight DNA contamination. When used to prepare a subset of samples for NGS, these quantity and quality measures produced libraries that passed clinical quality sequencing metrics (Figure 3C). By running qPCR for both 129 and 305 bp, we are more accurately able to discern quality of the sample through looking at the portion of sample that is greater than 129 and 305 bps. As cfDNA exhibits a narrow size distribution around 160-180 bp, 129-bp qPCR results accurately detect the total quantity of cfDNA and high molecular weight gDNA. 305-bp qPCR results indicate only the presence of high molecular weight cellular gDNA contamination.
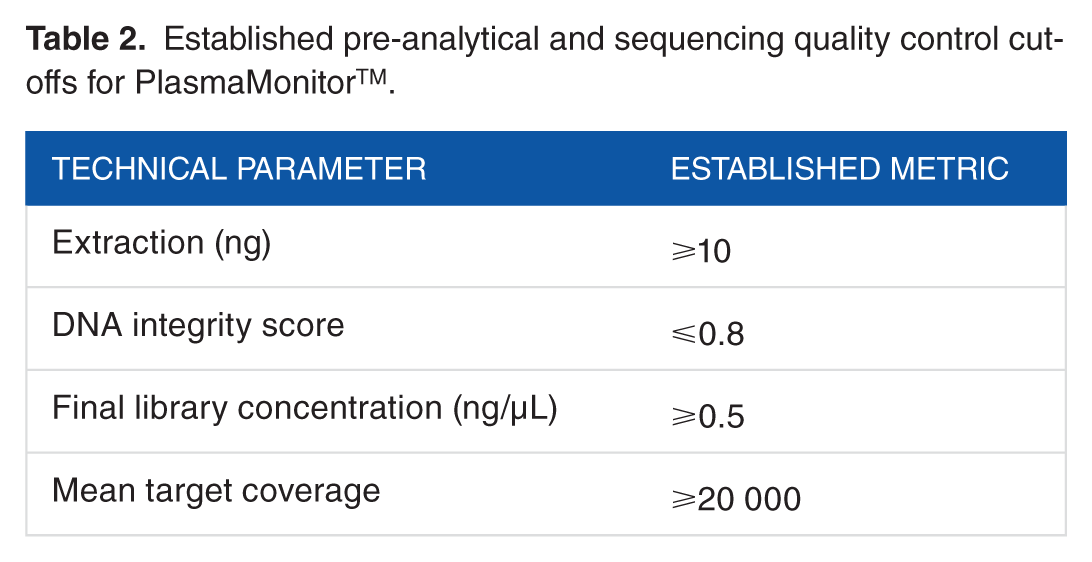
Validation for the PlasmaMonitorTM for clinical use included sequencing of 21 molecularly uncharacterized tumor samples, and 14 known controls under established metrics and was completed in 5 phases: (1) sample processing for validation parameter determination; (2) LOD, sensitivity, specificity, and accuracy; (3) inter-assay concordance; (4) intra-assay concordance; and (5) clinical validity in terms of interpretation and reporting of variants identified, per recommended guidelines30,31 (Figure 3D and E). The final clinical protocol was developed using an input of 10 ng of cfDNA quantified and qualified by a custom qPCR assay to establish DIN values, where ⩽0.8 was considered optimal (Table 2). Using samples containing variants with known allele frequencies and ddPCR-based confirmation of novel variants identified by our assay, we established the assay’s LOD.
Established pre-analytical and sequencing quality control cut-offs for PlasmaMonitorTM.
We used the SeraSeq ctDNA reference materials which contain a list of 11 hotspot cancer mutations at known allele frequency (1.2%, 0.5%, and 0.1%) in the following genes: AKT1, BRAF, EGFR, KRAS, PIK3CA, and TP53. In addition, we used three clinical samples with ddPCR-validated hotspot variants at 0.6%, 1%, and 5% allele frequency. Using these samples and the corresponding SeraSeq circulating tumor wild-type control, we designed a titration experiment to obtain samples with allele frequencies of 0.2%, 0.3%, 0.4%, 0.6%, 0.7%, 0.8%, 0.9%, and 1%. At each allele frequency from 0.1% to 1.2%, we then evaluated the assay’s ability to correctly identify known variants (sensitivity). At allele frequency of ⩾0.9%, we achieved a sensitivity of ~96% and we therefore deduced 0.9% as our assay’s LOD. At this LOD, sensitivity, specificity, and accuracy were found to be 96.6%, 100%, and 98%, respectively (Table 3). As the validation results indicate, the PlasmaMonitorTM is a well designed genetic test that is able to provide a comprehensive actionable report to the patient (Supplemental Figure 1).
PlasmaMonitor™ analytical performance characteristics and results.

Current Guidelines (Recommendations) for ctDNA Testing
A joint review assessing ctDNA testing in oncology was recently released by the American Society of Clinical Oncology (ASCO) and the College of American Pathologists (CAP). 32 This review surveyed the current status of ctDNA analysis in the field and provides a framework for the future development of ctDNA tests. The guidelines outline four areas of significance, namely, pre-analytical challenges, analytical validity, interpretation and reporting of variants, and the clinical validity and clinical utility (Table 4) to be specifically considered during the development of ctDNA assays. 32
Summary of ASCO/CAP (Merker, Oxnard and Compton, 2018) considerations for liquid biopsy assays in the clinic.

Receipt and processing of clinical samples has to follow the same established process (stringency and quality control) for each sample from beginning (accessioning) to end (sending the report out to the physician). Pre-analytical considerations 33 include optimal blood sampling, handling protocols, and long-term plasma and cfDNA storage which need to be considered in parallel with test validation criteria 32 (Figure 3D and E). Specimen handling from collection through shipping, transportation, and storage needs to be performed with the utmost care and adherence to regulations including chain of custody, while addressing sample stability since these aspects are critical and impact downstream processing.
Shipping and storage conditions of the plasma samples can cause varying concern as stability can decrease in a few days. To offset these variables, clinical testing laboratories including JAX provide a requisition form that includes acceptable conditions of samples to ensure that the collecting site is aware of the requirements prior to sample collection. It has been shown that improper shipping and storage conditions may lead to damage of DNA nucleases resulting in genomic DNA contamination. An additional aspect of specimen collection is the blood draw site, as no data are currently available comparing blood acquisition from other sites compared with peripheral veins, and phlebotomists should follow the tube manufacturer’s instructions for use. 32
Plasma is stable when stored at −80°C for long-term storage. While using plasma, attention should be paid to the type of tubes used for blood collection. Stability of cfDNA can be increased using EDTA since it inhibits nuclease (DNAse I) activity; heparin, however, has been shown to impact downstream processing of plasma including yield of cfDNA obtained. Condition of specimen is also equally important, particularly in terms of hemolysis, which can spuriously increase DNA yields. However, blood storage tubes incorporating EDTA, have a bigger incidence rate of genomic DNA (gDNA) contamination if blood is stored in them for more than 7 days. gDNA increased ~2-fold on day 14 in blood collection tubes (BCT) tubes (Streck) as opposed to a 456-fold increase in K3EDTA tubes, 34 making them the preferred tubes for specimen collection for liquid biopsies. Upon receipt of the blood sample at JAX, to ensure proper sample processing and downstream analysis of the PlasmaMonitorTM, plasma is immediately isolated and stored in the −80°C freezer to maintain stability and avoid degradation and contamination of DNA.
Analytical validity is essential and needs to be clearly established for all clinical grade assays. Currently, the ASCO/CAP consensus for cfDNA assays concludes that there is a discordance between solid tumor sequencing and liquid biopsy testing and therefore suggests that solid tumor evaluation be followed by a liquid biopsy assay for concordance and recommend the confirmation of variants identified in liquid biopsy assays by orthogonal methods. 32 A critical aspect of the difference between solid tumor and plasma-based evaluation is the LOD and VAF. Defining LOD for an assay is of utmost importance, for ctDNA assays in particular, the LOD will be lower than those of solid tumor genotyping assays due the small amounts of DNA being shed from the tumor.
Similar to FoundationOne® Liquid panel (Table 1), which can act as a stand-alone panel or accompany the FoundationOne CDx™ FDA-approved solid tumor panel, the PlasmaMonitorTM is established to be used for the evaluation of both solid tumor as well as plasma samples. The PlasmaMonitorTM has a LOD of 0.9%, which complements our solid tumor assay and allows for variant detection as well as confirmation, if needed. We do note that there are other liquid biopsy tests currently available such as the Guardant360 which can detect very low-frequency variants (0.01%), in contrast to the PlasmaMonitorTM. For those variants identified in our solid tumor evaluation that are not present in the PlasmaMonitorTM, we have developed and validated a ddPCR assay. In line with the shortcomings acknowledged by the ASCO/CAP consensus panel, we approached the concern of the liquid biopsy test being less reliable when compared with solid tumor testing, in terms of the high false-positive rate, by orthogonally confirming mutations detected by PlasmaMonitorTM using ddPCR, prior to reporting detected gene variants. We have also restricted analysis to only three nucleotide positions within a given hotspot for effective variant detection. False-negative results still remain a challenge and continued optimization efforts are ongoing to ensure that the PlasmaMonitorTM can accurately act as a confirmation assay to deliver the most accurate information to the patients we serve. With regard to performance of monitoring setting across a subset of clinical samples in different time points, we predict and expect that samples regardless of time points pass all set QC thresholds for the assay.
The ASCO/CAP review also raises a concern regarding the issue of discerning between analytical and clinical validity of liquid biopsies. Clinical validity including quantifying tumor origin and minimum residual disease leads the article to conclude that there is only emerging evidence of clinical validity in this application. 32 For the PlasmaMonitorTM, our intent would be to monitor the detection of variants being directly targeted for therapy or the emergence of well-established resistance mutations in the patient. Our validation included appropriate dilution experiments to stratify detection along a variety of ranges and the operational LOD cut-off was chosen above theoretical LOD to minimize false-positive results (Figure 3D). We agree that there are serious limitations, both technical and interpretive with the analytical and clinical validity. We therefore make zero claims or recommendations regarding such relationships, rather, only focusing on them as limitations of the assay.
In addition to clinical validity, clinical utility is yet to be demonstrated for the evaluation of cfDNA to monitor disease. To combat the absence of clinical utility on its own, physicians receiving results from our PlasmaMonitorTM have access to a multidisciplinary genomic tumor board to discuss the utility of the reported results (Supplemental Figure 1) on a case-by-case basis thus providing a method of clinical utility for our patients. For the reasons above, we believe we have employed the assay as responsibly as can be done with current research/knowledge/evidence while addressing the needs and demands of a diverse patient and physician population.
Challenges and Limitations of Liquid Biopsy Assays
Although liquid biopsy has the potential to be an effective assay in the detection of clinically relevant variants, monitoring the progression of cancer via evaluating exosomes, CTCs or cfDNA, can be challenging due to multiple factors including but not limited to tumor stage, tumor type, tumor heterogeneity, and tissue of origin, necessitating the need for high sensitivity and specificity and increased robustness of current technologies. Because cfDNA is a mix of normal DNA as well as non-mutated tumor DNA in addition to tumor DNA, separating out enough ctDNA can prove to be a challenge depending on tumor type and stage.
Allelic frequencies and tumor heterogeneity contribute to the false-negative rate. In early-stage tumors especially, genomic alterations have been known to be present at very low variant allele frequency (VAF) which increases the sensitivity and the specificity 35 needed for the assay to detect these variants. 36 ctDNA used for genomic variant genotyping represents an extremely small fraction of the tumor, in some cases only less than 0.01%. 37 In parallel, CTCs in early-stage tumors commonly occur at extremely low frequencies, making detection and recovery of the individual cells a burdensome process. Detection of CTCs requires a greater volume of blood from the patient or highly sensitive and specific analytic methods with complex enrichment steps to receive optimal results.12,38,39 CTCs and cfDNA both when targeted, sequenced, and molecularly characterized might only represent a subpopulation of the tumor in the intra-tumor heterogeneity landscape. 12 Aiming to capturing the full heterogeneity of the tumor with ctDNA presents the possibility of the presence of DNA from normal cells. This leaves room for misinterpretation of the tumor because of the background presence of leukocytes and the overlap they may have with cancer cells for the genes of interest. 40 Specimen quality, short half-life of CTCs, and circulating nucleic acids as well impact the final results. The PlasmaMonitorTM was developed to follow mutations detected in paired solid tumor/plasma samples at low VAF and LOD, with the aim to reduce false-negative results; however, stage of cancer and tumor heterogeneity could yet contribute to the false-negative calls of the assay.
Discussion and Conclusions
In conclusion, liquid biopsy has the advantage of being a minimally invasive or non-invasive quick method for the detection and analysis of circulating biomarkers, enabling us to evaluate a patient at multiple time points during disease management. However, given the absence of clinical utility and the need for highly sensitive robust technologies to better address the false-positive and false-negative rates, the liquid biopsy assays are still in their infancy, leaving the tissue biopsy as the superior method of targetable mutation identification across a majority of different tumor types. The JAX PlasmaMonitorTM provides detection of 84 hotspot SNVs and indels across 14 clinically actionable genes to provide relevant clinical information to both the treating physician and the patient, post-solid tumor testing, as a way to follow known mutations and monitor disease. Given the promise of this area of precision medicine, there is clearly a focused effort in driving the current technologies to address this unmet need of the patient, with the goal to make liquid biopsy a surrogate to traditional methods of tumor profiling to play a significant role in the early detection, prognostication, and therapy guidance.
Author Contributions
All authors contributed to generation and evaluation of data and manuscript writing. HR was responsible for conception, design and critical editing of the manuscript for this study. All authors approved the final manuscript.
Supplemental Material
Supl_Figure_1_xyz133377e7940cc – Supplemental material for Technical and Regulatory Considerations for Taking Liquid Biopsy to the Clinic: Validation of the JAX PlasmaMonitorTM Assay
Supplemental material, Supl_Figure_1_xyz133377e7940cc for Technical and Regulatory Considerations for Taking Liquid Biopsy to the Clinic: Validation of the JAX PlasmaMonitorTM Assay by Bridgette A Sisson, Jasmina Uvalic, Kevin Kelly, Pavalan Selvam, Andrew N Hesse, Guruprasad Ananda, Harshpreet Chandok, Daniel Bergeron, Lauren Holinka and Honey V Reddi in Biomarker Insights
Footnotes
Acknowledgements
The authors gratefully acknowledge Jane Cha (JAX Creative Design) for the graphics and schematic presented in Figure 1. BAS and JU have equal contribution.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
This publication did not involve the use of patient data.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.