
Abstract
Background & aim:
Oral squamous cell carcinoma (OSCC) is a devastating disease with poor prognosis and low survival rates, despite advancements in diagnosis and treatment. Early detection and identification of molecular targets are crucial for improving patient outcomes. This study aims to identify differentially expressed genes (DEGs) and key molecular pathways involved in the OSCC. This study’s findings will contribute to the development of effective targeted therapies, ultimately improving the prognosis and survival rates of OSCC patients.
Materials & methods:
Three gene expression profiles (GSE37991, GSE30784, and GSE107591) from the GEO database were analyzed for differentially expressed genes using EnrichR. Subsequent downstream analyses of the selected module genes were conducted using various bioinformatics tools including STRING, Cytoscape, GEPIA, cBioPortal, NetworkAnalyst, MirWalk, and a bipartite miRNA-mRNA correlation network.
Result:
The reanalysis indicated that the Toll-like receptor (TLR) signaling pathway plays a significant role in the development of oral SCC and CXCL8, CCL5, CXCL10, STAT1, IL1B, and TLR2 genes were up-regulated and enriched significantly in the signaling pathways’ interactions in oral SCC. Genetic mutation analysis of hub genes in OSCC revealed that STAT1 have 2.5% mutation rate and 0% for other genes. It was revealed that the development and prediction of OSCC may be affected by hsa-mir-146a-5 and hsa-mir-155-5p.
Conclusion:
Novel potential biomarkers and signaling pathways associated with OSCC have been identified, which may be important in the transformation of OSCC adenocarcinoma and may serve as therapeutic targets for OSCC.
Keywords
Highlights
Identification of 6 hub genes associated with oral cavity squamous cell carcinoma.
Toll-like receptor signaling pathway was significantly enriched.
STAT1, CXCL8, CXCL10, IL1B, TLR2, and CCL5 were upregulated.
Potential involvement of hsa-miR-146a-5p and hsa-miR-155-5p in OSCC pathogenesis.
Introduction
Oral squamous cell carcinoma (OSCC) can arise from various regions of the oral cavity, including the lips, tongue, palate, buccal mucosa, gums, floor of the mouth, vestibule, and retromolar region. 1 OSCC represents a major global health challenge due to its aggressive biological behavior and poor prognosis. 2 Despite advancements in diagnostic and therapeutic techniques, the survival rate of patients with OSCC remains low. 3 Early detection of oral cancer, including its precancerous stages, significantly improves patient outcomes. Benign oral cavity lesions commonly occur in the anterior tongue, buccal mucosa, floor of the mouth, hard palate, retromolar trigone, and gingiva. 4 Globally, oral cancer accounts for approximately 200 000 new cases and 100 000 deaths annually. The global age-standardized prevalence of lip cancer in 2012 was 0.3 per 100 000 people (0.4 in men and 0.2 in women). 5 Several etiological factors contribute to the development of OSCC, including human papillomavirus (HPV) infection, tobacco smoking, alcohol consumption, and betel nut chewing. 6 Surgery remains the primary treatment modality for OSCC, with the choice of surgical technique depending on the tumor’s anatomical location, extent of invasion, margins, and histopathological characteristics. 7 However, the modest improvement in the 5-year overall survival rate from 63% to 65% over the past 8 years highlights the limited impact of recent clinical advances on OSCC prognosis. 8 Therefore, deciphering the molecular mechanisms underlying OSCC development and progression is crucial for identifying effective therapeutic targets. 3 Comprehensive molecular studies are needed to elucidate the key pathways and biomarkers involved in OSCC pathogenesis.
The present study aims to address these gaps by integrating multiple open-access gene expression datasets to identify differentially expressed genes (DEGs) and the major molecular pathways associated with OSCC. By combining data from several high-quality sources, this study provides a more comprehensive and accurate molecular profile of OSCC. Rigorous selection criteria were applied to ensure data reliability and relevance. Through integrated bioinformatics analysis, this approach minimizes dataset-specific bias and enables a robust identification of DEGs and key signaling pathways. Furthermore, survival and mutation analyses of the identified hub genes offer deeper insights into their prognostic and functional roles in OSCC progression.
Materials and Methods
Data Collection
The Gene Expression Omnibus (GEO) database 9 contains numerous gene expression datasets; however, only a subset is suitable for identifying hub genes associated with OSCC tumor progression. To ensure data relevance and reliability, the following inclusion criteria were applied:
Samples must originate from human subjects.
Datasets must be analyzable using the GEO2R online tool.
Each dataset must include more than 6 samples.
Both normal and OSCC (disease) groups must be available for comparison.
All samples must represent oral cavity tumor tissues.
Based on these criteria, 3 gene expression datasets (GSE37991, GSE30784, and GSE107591) were retrieved from the NCBI-GEO database (https://www.ncbi.nlm.nih.gov/geo/) the platform identifiers for GSE30784, GSE37991, and GSE107591 were GPL570, GPL6883, and GPL6244, respectively. These datasets were selected to identify key molecular targets and pathways potentially involved in OSCC. Among the available OSCC-related GEO datasets, GSE37991, GSE30784, and GSE107591 were selected because they fulfilled all predefined inclusion criteria and provided high-quality expression profiles with sufficient sample sizes. The datasets were generated using well-established microarray platforms (GPL570, GPL6883, and GPL6244) and contained both OSCC and normal oral tissue samples. To avoid potential batch effects caused by platform or sample heterogeneity, each dataset was normalized individually, and only overlapping DEGs consistently identified across all 3 datasets were included for downstream analysis. This intersection-based approach ensured analytical robustness and minimized cross-platform bias.
Data Processing of DEGs
Differentially expressed genes (DEGs) between normal and OSCC samples were identified using the GEO2R online analysis tool (https://www.ncbi.nlm.nih.gov/geo/geo2r/). Expression data were normalized by quantile normalization prior to analysis. Genes with an adjusted P-value <.01 and |log FC| > 1 were considered significantly differentially expressed. Identified DEGs were categorized as upregulated or downregulated based on fold-change values. Overlapping DEGs among the 3 datasets were determined using a web-based Venn diagram tool (http://bioinformatics.psb.ugent.be/cgi-bin/liste/Venn/calculate_venn.htpl).
Enrichment Analysis
To explore the biological significance of the DEGs, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed. These analyses identified key biological processes (BP), molecular functions (MF), and cellular components (CC) potentially associated with OSCC. Functional enrichment was conducted using the Enrichr web server10 -13 and the Database for Annotation, Visualization, and Integrated Discovery (DAVID; https://david-d.ncifcrf.gov). Significance was defined as an adjusted P-value <.0514,15 Enrichr was also applied to annotate genes within specific modules identified from network analyses.
Protein‒protein Interaction (PPI) Network and Module Analysis
The Search Tool for the Retrieval of Interacting Genes (STRING; https://string-db.org/) was used to construct a PPI network of the DEGs, with a minimum required interaction score > 0.4.16,17 The network was visualized and analyzed using Cytoscape version 3.7.2. 18 Highly interconnected gene clusters (modules) were identified using the Molecular Complex Detection (MCODE) plugin with the following parameters: degree cutoff = 2, node score cutoff = 0.2, k-core = 2, and maximum depth = 100. Hub genes were determined using the CytoHubba plugin based on node degree and network connectivity. 19 Four topological analysis algorithms (Degree, MCC, MNC, and EPC) were applied in CytoHubba to comprehensively evaluate gene centrality from different perspectives. The intersection of the top-ranked genes across these methods was defined as the final set of hub genes, ensuring robust and consistent selection.
Survival Analysis of Hub Genes
The prognostic significance of hub genes was evaluated using the Kaplan–Meier Plotter tool. In addition, the Gene Expression Profiling Interactive Analysis (GEPIA) web server. 20 Which integrates data from TCGA and GTEx, was used to assess expression differences between OSCC and normal tissues. The log-rank test P-value, hazard ratio (HR), and 95% confidence interval (CI) were calculated for each hub gene.
Mutation Analysis of Hub Genes
To identify genetic alterations in hub genes, mutation data were analyzed using the cBioPortal database21,22 this platform provides comprehensive OSCC genomic data, including whole-genome sequencing results from 40 OSCC tumors and matched normal tissues. 23
Potential miRNA‒mRNA Interactions
Potential interactions between differentially expressed miRNAs (DEmiRs) and hub genes were predicted using the miRWalk database. The overlap between miRNA target genes and module genes was visualized using a Venn diagram. A bipartite miRNA–mRNA regulatory network was constructed and analyzed using Cytoscape software. 24
Results
Identification of DEGs
These 3 profiles included 101 samples of normal oral mucosa and 224 samples of OSCC (Table 1). Three gene expression datasets, GSE30784, GSE37991, and GSE107591, were selected from the GEO-NCBI for OSCC after the DEGs between OSCC and normal oral mucosa were screened. For all analyses, adjusted P-values <.01 and |log2FC| > 1 were taken into account. There was an overlap across the 3 datasets, depicted by a Venn diagram showing 1157 downregulated DEGs and 941 upregulated DEGs (Table 2 and Figure 1A and B).
The Details of the GEO Datasets.

Venn Diagram Results from 3 GEO Datasets for SCC.


Identification of common differentially expressed genes (DEGs) across 3 OSCC datasets. (A) Venn diagram of upregulated DEGs identified from GSE30784, GSE37991, and GSE107591 datasets. (B) Venn diagram of downregulated DEGs from the same datasets. The overlapping regions represent genes consistently up- or downregulated across all 3 datasets. A total of 943 upregulated and 1159 downregulated DEGs were found to be shared, which were used for subsequent functional and network analyses.
Functional Enrichment Analysis
Functional enrichment was examined using Enrichr, a feature-rich web service for the analysis of gene set enrichment. The KEGG pathways were assessed to specify the significant pathways associated with the DEGs. Based on KEGG analysis, the DEGs for SCC were both upregulated and downregulated (Table 3).
KEGG in SCC.

PPI and Modular Analysis and the Identification of Hub Genes
A total of 1878 genes 13 602 edges in OSCC were detected as differentially expressed genes in OSCC, utilizing Cytoscape 3.8.0 and the STRING 11.0 database. Afterward, the resulting PPI network was evaluated. The uppermost 20 genes were determined by the techniques of Degree, MNC, EPC, and EPC using the CytoHuuba plugin in Cytoscape software. Twenty-four hub genes associated with OSCC were identified via the Venn diagram (Table 4). Seventeen genes were upregulated; 7 were downregulated.
The Top Core Genes Screened in 0SCC Based on the Degree/MCC/MNC/EPC.

Hub Gene Enrichment
KEGG pathway enrichment analysis was performed to identify the key genes connected to SCC. The top-ranked genes identified in our analysis are considered hub genes central to the development of SCC. The reanalysis indicated that the Toll-like receptor (TLR) signaling pathway plays a significant role in the development of oral SCC and CXCL8, CCL5, CXCL10, STAT1, IL1B, and TLR2 genes were up-regulated and enriched significantly in the signaling pathways’ interactions in oral SCC (P < .05, Figure 2).

Analysis of the top genes SCC by KEGG pathway enrichment.
Expression Analysis of Core Genes in SCC
The 6 hub genes’ expression identified in the core gene analysis was validated using the GEPIA database (P < .05, Figure 3).

Differentially expressed 6 hub genes in OSCC patients, compared to healthy controls according to the GEPIA online database (P < .05).
Prognosis and Survival Rates of the Core Genes in OSCC
The Kaplan-Meier plotter was utilized to evaluate the prognostic significance of the top genes associated with OSCC, determining their relation to low survival outcomes in OSCC patients (Figure 4).

The Kaplan-Meier plotter used to determine the prognostic value of 10 core genes in SCC related to the survival rate.
Mutational Analysis of Hub Genes Involved in OSCC
Signal transducer and activator of transcription 1 (STAT1) was the most significant gene linked to amplification mutations according to a study’s findings performed in 2013 25 on hub genes involved in OSCC conducted by MD Anderson, Cancer Discov, which involved the mutational analysis of 40 samples (Figure 5).

Genetic mutation analysis of hub genes in OSCC. The mutation rate is 2.5% for STAT1 and 0% for other genes.
Bipartite miRNA-mRNA Network Analysis
The mirWalk database predicted potential microRNAs, possibly interacting with specific hub genes (CXCL8, CCL5, CXCL10, STAT1, IL1B, and TLR2) to examine their function in the development of OSCC. Additionally, the miRTarBase filter was taken into consideration. Four miRNAs that are significant in OSCC were found by evaluating the shared miRNAs among these genes (Table 5 and Figure 6).
The miRNAs Involved in the Hub Genes.

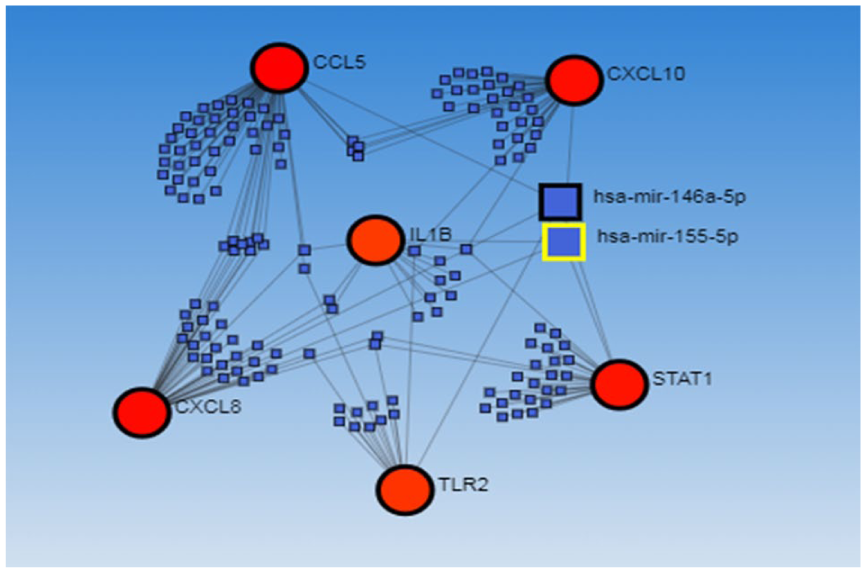
Bipartite miRNA‒mRNA regulatory network of important miRNAs involved in SCC.
Validation of the Key Candidate DEGs
To analyze cancer transcriptome data, UALCAN (http://ualcan.path.uab.edu/index.html) is utilized. The expression of several genes in various tumor types and the correlation between genes and prognosis may be obtained by UALCAN through comprehensive studies of TCGA gene expression data 12 The UCLAN database received the potential important genes, and the association between the expression of these genes and the prognosis of OSCC was confirmed using the TCGA data (accession date: November 21, 2018).
Discussion
Oral squamous cell carcinoma (OSCC) is among the most prevalent types of head and neck squamous cell carcinoma (HNSCC) 1 Although OSCC is characterized by high mortality, aggressive invasion, and rapid metastasis, reliable biomarkers and therapeutic targets for early diagnosis and effective treatment remain limited. Identifying genes differentially expressed between OSCC and normal tissues can provide valuable insights into the underlying molecular mechanisms and lead to the discovery of potential diagnostic and therapeutic targets.
Advancements in microarray and high-throughput sequencing technologies have enabled large-scale genomic analyses, offering new opportunities to understand the molecular basis of OSCC. This comprehensive bioinformatics study aimed to integrate multiple datasets to overcome the limitations of individual studies and provide a more robust and accurate molecular landscape of OSCC. By combining data from multiple populations and platforms, this analysis contributes to improving our understanding of OSCC pathogenesis and supports the development of new diagnostic and therapeutic strategies.
In this study, 3 OSCC gene expression datasets comprising 224 tumor samples and 101 normal controls were analyzed. Venn diagram analysis identified 941 upregulated and 1157 downregulated differentially expressed genes (DEGs) common to all datasets. Subsequent bioinformatics analyses—including PPI network construction, enrichment analysis, module identification, and hub gene selection—highlighted several key genes and pathways potentially involved in OSCC progression. The Toll-like receptor (TLR) signaling pathway, in particular, appeared to play a crucial role, and 6 hub genes (CXCL8, CCL5, CXCL10, STAT1, IL1B, and TLR2) were identified as key molecular markers.
Because gene expression can vary across populations, our inclusion of datasets from the USA, Taiwan, and Italy helped minimize sample heterogeneity and identified DEGs consistently associated with OSCC across different ethnic backgrounds. This cross-population validation strengthens the robustness and potential generalizability of our findings.
Several previous bioinformatics studies have explored molecular alterations in OSCC.5,6 For instance, Xu et al 26 identified 651 DEGs enriched in cytokine–cytokine receptor interaction and xenobiotic metabolism pathways suggesting potential biomarker roles for CSF2 and EGF. Similarly, Zou et al 27 identified 34 DEGs across 4 datasets, emphasizing SPP1 and PLAU in OSCC cell migration and proliferation. Mathavan et al 7 reported 9 hub genes, including APP and EHMT1, associated with OSCC and demonstrated that fisetin may regulate these genes. Compared with these studies, our analysis integrated 3 independent datasets to reveal 6 hub genes highly consistent across populations and biologically relevant to inflammation and tumor immune responses, thus providing broader and more reproducible results.
The hub genes identified in our study play important roles in inflammation, immune modulation, and tumor progression. For example, CXCL8 and CXCL10, both members of the chemokine family, have been shown to promote cancer development and predict patient outcomes by mediating pro-inflammatory and pro-oncogenic signaling28,29 Elevated CXCL8 levels in endothelial–tumor cell co-cultures suggest its involvement in promoting tumorigenesis 30 TLR2, another identified hub gene, is known to regulate immune responses in OSCC and other epithelial malignancies. While TLR2 activation on inflammatory cells may promote antitumor immunity, its overexpression in tumor keratinocytes can enhance resistance to apoptosis and favor tumor survival31 -34 Furthermore, STAT1 is widely recognized for its role in inducing apoptosis and cell cycle arrest through immune-mediated signaling, and its activation status has been linked to OSCC prognosis35,36 Similarly, IL1B, a pro inflammatory cytokine, contributes to extracellular matrix degradation and OSCC invasiveness by promoting MMP production.37,38 Lastly, CCL5 promotes tumor invasion and migration by interacting with its receptor CCR5 and upregulating MMP-9 expression in oral cancer cells.39 -42
Together, these findings underscore the importance of immune and inflammatory pathways—particularly chemokine signaling, STAT1-mediated apoptosis, and TLR activation—in the pathogenesis and progression of OSCC. Targeting these pathways may provide new opportunities for therapeutic intervention.
Despite these promising findings, several limitations should be acknowledged. First, our study primarily relied on publicly available transcriptomic datasets and bioinformatics analyses. Experimental validation, such as in vitro and in vivo functional assays, is still required to confirm the biological roles of the identified hub genes. Second, although integrating datasets from different populations reduced heterogeneity, potential batch effects and sample biases cannot be completely ruled out. Third, only 6 hub genes were investigated in depth; other DEGs may also play important roles and warrant further exploration. Additionally, the mutation analysis was based on 40 OSCC samples from the MD Anderson 2013 dataset. The relatively small sample size may limit the accuracy of estimated mutation frequencies, and these findings should therefore be interpreted with caution. Finally, a larger clinical cohort is needed to validate the diagnostic and prognostic utility of these hub genes in independent patient populations. The hub genes and pathways identified in this study, including CXCL8, CCL5, CXCL10, STAT1, IL1B, and TLR2, may serve as potential biomarkers for OSCC diagnosis or prognosis. Their involvement in immune and inflammatory signaling suggests they could also represent therapeutic targets, particularly for interventions aimed at modulating the tumor microenvironment. While these findings are currently based on bioinformatics analyses, they provide a foundation for future experimental studies and clinical validation, which could ultimately support the development of targeted therapies or predictive biomarker panels in OSCC patients. Future research should therefore focus on experimental and clinical validation to translate these findings into practical clinical applications.
Conclusion
Using integrated bioinformatics analysis, this study identified 941 upregulated and 1157 downregulated differentially expressed genes (DEGs) across 4 OSCC datasets. These DEGs were mainly associated with Toll-like receptor (TLR) and chemokine signaling pathways. Six hub genes—CXCL8, CCL5, CXCL10, STAT1, IL1B, and TLR2—were highlighted as key molecules potentially involved in OSCC pathogenesis. While several of these genes, such as STAT1 and CXCL8, have been previously reported to contribute to OSCC progression, our integrated analysis reinforces their importance across multiple independent datasets and provides additional network- and pathway-level insights. These results enhance the current understanding of the molecular mechanisms underlying OSCC and may assist in identifying biomarkers or therapeutic candidates worthy of further validation. Nevertheless, additional experimental and clinical studies are required to confirm the functional roles of these genes and to explore their potential translational applications in OSCC diagnosis and therapy.
Footnotes
Acknowledgements
The authors thank to the Tehran University of Medical Science for their financial support.
Author’s Note
Ethical Considerations
This study was conducted using publicly available datasets and in accordance with relevant ethical guidelines and regulations. Ethical approval was obtained from the research Ethics Committees of Amiralam Hospital Tehran University of Medical Sciences (IR.TUMS.AMIRALAM.REC.1402.011).
Consent to Participate
The study used publicy available datasets; therfore, informed consent was not required.
Consent for Publication
Written informed consent was obtained from all participants included in the study.
Author Contributions
MR. E is a PhD candidate in Cancer Research Institute, Tehran University of Medical Sciences, Tehran, Iran, and he was involved in writing-original draft, Visualization, Validation, Software, Resources, Methodology, Investigation, Formal analysis, Data curation, Conceptualization with support from M.A, E.K that were supervised the total of this project and monitoring all processes of function as the head of the team in collaboration with M.VR. SH.EM and SF.MH were two researchers that investigated the summary of papers for extraction of the data and quality evaluation. H.M and A.MK was involved in review & editing. All authors contributed to the article and approved the submitted version.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was financially supported by the Amiralam Hospital Tehran University of Medical Sciences.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.