
Abstract
Objectives:
Aim of this study was to explore the immune-related lncRNAs having prognostic role and establishing risk score model for better prognosis and immunotherapeutic coherence for esophageal cancer (EC) patients.
Methods:
To determine the role of immune-related lncRNAs in EC, we analyzed the RNA-seq expression data of 162 EC patients and 11 non-cancerous individuals and their clinically relevant information from the cancer genome atlas (TCGA) database. Bioinformatic and statistical analysis such as Differential expression analysis, co-expression analysis, Kaplan Meier survival analysis, Cox proportional hazards model, ROC analysis of risk model was employed.
Results:
Utilizing a cutoff criterion (log2FC > 1 + log2FC < −1 and FDR < 0.01), we identified 3737 RNAs were significantly differentially expressed in EC patients. Among these, 2222 genes were classified as significantly differentially expressed mRNAs (demRNAs), and 966 were significantly differentially expressed lncRNAs (delncRNA). Through Pearson correlation analysis between differentially expressed lncRNAs and immune related-mRNAs, we identified 12 immune-related lncRNAs as prognostic signatures for EC. Notably, through Kaplan-Meier analysis on these lncRNAs, we found the low-risk group patients showed significantly improved survival compared to the high-risk group. Moreover, this prognostic signature has consistent performance across training, testing and entire validation cohort sets. Using ESTIMATE and CIBERSORT algorithm we further observed significant enriched infiltration of naive B cells, regulatory T cells resting CD4+ memory T cells, and, plasma cells in the low-risk group compared to high-risk EC patients group. On the contrary, tumor-associated M2 macrophages were highly enriched in high-risk patients. Additionally, we confirmed immune-related biological functions and pathways such as inflammatory, cytokines, chemokines response and natural killer cell-mediated cytotoxicity, toll-like receptor signaling pathways, JAK-STAT signaling pathways, chemokine signaling pathways significantly associated with identified IRlncRNA signature and their co-expressed immune genes. Furthermore, we assessed the predictive potential of the lncRNA signature in immune checkpoint inhibitors; we found that programed cell death ligand 1 (PD-L1; P-value = .048), programed cell death ligand 2 (PD-L2; P-value = .002), and T cell immunoglobulin and mucin-domain containing-3 (TIM-3; P-value = .045) expression levels were significantly higher in low-risk patients compared to high-risk patients.
Conclusion:
We believe this study will contribute to better prognosis prediction and targeted treatment of EC in the future.
Keywords
Introduction
Despite the progress in modern techniques for detecting and treating esophageal cancer (EC), the overall prognosis for EC patients remains notably poor, marked by a high rate of recurrence. 1 Managing this type of cancer presents a challenge due to its inherent heterogeneity, leading to variable responses to similar treatments among different patients. As a result, there is a critical need for reliable molecular markers in order to help clinicians make more educated judgments about appropriate treatment methods for individual EC patients. Such markers would enhance treatment effectiveness, improving patient outcomes and survival rates.
Recent research underscores the significance of the tumor microenvironment (TME) in the development and progression of various cancers. 2 The TME comprises tumor cells, tumor-associated normal epithelial cells, vascular cells stromal cells, extracellular matrix, and immune cells, playing crucial roles in the crosstalk between tumor and non-tumorous cells. 3 Studies have revealed that infiltrating immune cells in the TME play a substantial role in initiating and progressing various malignancies, including esophageal cancer.4,5 For instance, tumor-associated macrophages promote metastasis and regulate the immune system in the TME, 6 while dendritic cell infiltration is associated with a favorable clinical outcome. 7 On the contrary, the infiltration of regulatory T cells (Tregs) hinders the body’s natural immune response against tumors and is associated with an unfavorable prognosis. 8 CD8-positive T lymphocytes, on the other hand, are associated with a good prognosis in several cancers. 9 These infiltrating immune cells have crucial functions in the onset and advancement of cancer, exerting either positive or negative effects on the patient’s prognosis as a result of tumor heterogeneity and the presence of diverse populations of immune cells infiltrating the tumor. 10 Given the crucial role of the immune system in tumor formation, progression, and treatment, immunotherapy provides a unique opportunity to treat malignancies effectively. 11 Immunotherapy amplifies the tumor microenvironment of antitumor immunity in order to eradicate cancer cells through the activation of the body’s immune system. 12 Immune evasion is closely related to the interaction between the tumor microenvironment (TME) and tumor cells. 5 Some studies suggest a positive correlation between the infiltration level of CD8-positive T-cells, B-cells, and macrophages with immune checkpoint inhibitors such as Programed cell death 1 (PD-1), Lymphocyte activation gene 3 (LAG3), programed cell death ligand 1 (PD-L1), T cell immunoglobulin and mucin-domain containing-3 (TIM3), and cytotoxic T-lymphocyte associated protein 4 (CTLA-4) expression.13 -17 Immunotherapy, including immune checkpoint inhibitors (ICIs), tumor vaccines, chimeric antigen receptor T-cell (CAR-T) therapy, and NK-cell-based therapy, has provided new hope for EC patients. 13 However, only a small percentage of patients benefit clinically from immunotherapy due to primary or acquired resistance. Therefore, a comprehensive understanding of the mechanisms by which EC cells evade antitumor immunotherapy is essential for a more effective multidisciplinary treatment strategy.
Recent research has shown that long non-coding RNAs (lncRNAs), longer than 200 nucleotides, are essential for several stages of cancer immunology, including antigen presentation, immunological activation, and immune cell infiltration. 18 Indeed, immune checkpoint inhibitor expression was negatively correlated with the lncRNA signature-based risk index, implying that the lncRNA signature may play a role in EC immunotherapy. 19 Recent research by our group and others has also shown that lncRNAs such as SNHG12, 20 LINC01133, 21 LINC00324, 22 TINCR, 23 CASC9, 24 EWSAT1 25 have the potential to function as prognostic and therapeutic biomarkers for various cancers. They regulate various hallmarks of cancers, including chromatin modification, transcription, and post-transcriptional processing.21,26 Pang et al identified an immune-related multi-lncRNA signature influencing esophageal cancer prognosis, utilizing transcriptome data from EC patients in both the TCGA and GEO databases. 27 Furthermore, an independent biomarker for the prognosis and the evaluation of ESCC’s response to immunotherapy using immune checkpoint inhibition was established by Zhu et al through the development of an 8-lncRNA signature-based risk model. 19 However, these studies lack the clinical implications of these lncRNA signatures on larger cohorts. Thus, based on the complexity and heterogeneity of the tumor microenvironment and the significance of lncRNAs, we analyzed RNA sequencing data from TCGA and immune-related mRNA from the ImmPort database. Twelve immune-related prognostic lncRNA signatures, namely AC008687.2, AC134312.5, AL031587.3, AL353764.1, HAND2-AS1, LINC00261, LINC01503, LINC01614, LINC02561, MYOSLID, TM4SF19-AS1, U62317.3, were identified through Pearson correlation in EC. Using these lncRNAs, we developed a predictive prognostic risk model to distinguish between high-risk and low-risk EC patients. Furthermore, we designed an integrated analysis combining tumor microenvironment and lncRNAs, evaluating the predictive potential of the lncRNA signature in immune cells and checkpoint inhibitors, intending to provide valuable insights into developing more effective personalized immunotherapeutic strategies for EC patients.
Materials and Methods
Data download and preprocessing
The RNA-Seq and clinical data of 162 EC patients and 11 normal samples were downloaded using the “TCGAbiolinks” R package from the publicly available database, the cancer genome atlas (TCGA; https://portal.gdc.cancer.gov). Gene re-annotation was performed using the “ensembldb” R package. Immune-related gene lists were acquired from the immunology database and analysis portal (ImmPort), a publicly accessible repository of subject-level human immunology databases designed for translational and clinical research purposes (https://www.immport.org/). The RNAseq expression in the form of transcripts per million reads (TPM) of each individual was integrated with respective clinicopathological information such as vital status, overall survival time, age, gender, pathological stage, clinical stage, histological grade, smoking history, and alcohol consumption for preparing a matrix file using the “dplyr” package.
Differential expression analysis of total RNAs, lncRNAs, and miRNAs
To find both coding genes (mRNA) and non-coding (lncRNAs) transcripts differentially expressed in EC samples, first, we preprocessed the data, and later, we analyzed the data using the “DESeq2” package in RStudio software. The expression levels were normalized and transformed log2(x + 1) using raw count data and compared differentially expressed total RNAs, lncRNAs, and miRNAs between EC samples and normal samples, with a log2FC > 1 + log2FC < −1 and false discovery rate (FDR) <0.01. Spliced transcripts alignment to a reference (STAR)-count files were exported and used for differential gene expression analysis.
Identifications of immune-related mRNAs and lncRNAs
To identify differentially expressed immune-related mRNAs (deIRmRNA) in EC samples, as mentioned earlier, we downloaded the immune-related gene list from the ImmPort database (https://www.immport.org/). Then, to find out immune related mRNAs that are differentially expressed in EC patient samples, we overlapped the demRNAs obtained in previous analysis and checked for overlaps with ImmPort immune genes. Next, keeping in mind that immune-related lncRNAs (deIRlncRNAs) co-expressed with immune-related mRNAs, 28 we performed Pearson correlation analysis between lncRNAs and immune-related mRNAs that were differentially expressed in EC samples. Finally, the immune-related lncRNAs with significant correlation (P-value < .05) were considered for further study.
Identification of immune-related lncRNAs having significant prognostic value for EC
The Survival analysis of all immune-related lncRNAs using Kaplan-Meier survival analysis (KM curve) was performed to find the significant lncRNAs having a role in the prognosis of EC patients with P-value < .05. For plotting Kaplan-Meier curves, we used expression values of each lncRNA and divided the population into 2 groups that is less than the median and greater than the median. Finally, the lncRNAs whose expression level can significantly differentiate between the overall survival of the patients were considered as prognostic lncRNAs. We used the R-package “ggplot2” for plotting Kaplan–Meier curves.
Construction of immune-related lncRNA signature-based risk model
A personalized risk assessment model was developed for each individual patient to forecast the prognosis of esophageal cancer patients. This model incorporated the expression levels of specific immune-related lncRNAs that were identified as optimal prognostic markers. The estimated regression coefficients, obtained through Cox proportional regression analysis, were then multiplied by the expression levels of these lncRNAs to calculate the immune lncRNA signature-based risk score. The risk score was determined using the formula: Risk score = (β1)(X1) + (β2)(X2) + ... + (βn)(Xn), where β represents the regression coefficient and X represents the expression level of each prognostic immune-related lncRNA. Based on the median risk score, patients were categorized into low-risk and high-risk groups. To validate the effectiveness of this risk stratification, Kaplan-Meier survival analysis was conducted using the “survminer” package in R. This analysis aimed to determine if the high-risk and low-risk groups, established using the immune-related lncRNA-based risk score, exhibited significant differences in overall survival.
Validation of the immune-related lncRNA signature
Patients were allocated based on their individual risk scores, along with their clinical data. The cases were divided into high-risk and low-risk sets for further investigation using the median risk score. Subsequently, Kaplan-Meier survival curves were examined in both sets. To assess the predictive survival performance of our risk score, time-dependent receiver operating characteristic (ROC) curves were plotted using the “survivalROC” package. These curves were generated for the test group, train group, and the entire dataset. The “ggplot2” package was utilized for plotting these ROC curves.
Gene set enrichment analysis
To gain fresh perspectives on the role of the 12-lncRNA signature, we conducted an in-silico functional analysis to uncover potential biological functions and pathways linked to this signature in EC. For this, we used GSEA (https://www.gsea-msigdb.org/gsea/msigdb/human/annotate.jsp) 29 analysis to predict the enriched GO biological processes, cellular components, Molecular processes, and KEGG pathways. The top 10 GO terms and KEGG pathways were plotted against the number of overlapping genes and −log10(FDR) values.
Evaluation of immune cell infiltration and immune microenvironment
Tumor-infiltrating immune cells were already estimated for TCGA data by number of databases using algorithms such as TIMER, XCELL, CIBERSORT, MCPCOUNTER, ESTIMATE, and EPIC. So, we used the ESTIMATE algorithm to get the immune score, stromal score, and estimate score for individual EC patients. We downloaded the profile of infiltration estimation from the CIBERSORT database to explore the relationship of Immune-related lncRNA based risk score and infiltration of tumor-infiltrating immune cells in esophageal cancer.
Association of risk score and immune checkpoints
Previous studies suggest that immune checkpoint blockade therapy-related essential gene expression may be connected to immune checkpoint blockade treatment responsiveness. 15 We utilized a set of 8 crucial genes associated with immune checkpoint inhibitors therapy: programed death ligand 1 (PD-L1, also referred to as CD274), programed death 1 (PD-1, also known as PDCD1), T-cell immunoglobulin domain and mucin domain-containing molecule-3 (TIM-3, also known as HAVCR2), cytotoxic T-lymphocyte antigen 4 (CTLA-4), programed death ligand 2 (PD-L2, also known as PDCD1LG2), Lymphocyte-activation gene 3 (LAG3), V-domain Ig suppressor of T cell activation (VISTA) also known as VSIR, and T cell immunoreceptor with Ig and ITIM domains (TIGIT). Our objective was to explore the underlying significance of our immune-related lncRNA signature-based risk score in the immune checkpoint blockade treatment of patients with EC by examining the correlation between our Risk score and the expression levels of these critical genes.
Statistical analysis
The statistical analyses in this study were conducted using R version 4.0.2 and the relevant packages. For the Kaplan-Meier survival analysis, the “survminer” package was utilized, and the resulting graphs were generated using the “ggsurvplot” function from the “ggplot2” R-package. To assess the prognostic accuracy of the immune lncRNA signature, a time-dependent receiver operating characteristic (ROC) analysis was performed using the “survivalROC” package. Student’s t-test was employed for analyzing continuous variables, while ANOVA was used to examine the association between immune lncRNA expression and clinicopathological characteristics. Pearson correlation analysis was conducted to determine the correlation between variables. Additionally, multivariate regression analysis was carried out using the Cox proportional hazards model. Variables with P-values < .05 in univariate analyses were included in subsequent multivariate analyses. Graphs were plotted using both the “ggplot2” R-package and graph pad prism 8.0.1.
Results
Coding mRNAs and long non-coding RNAs are differentially expressed in EC
To identify differentially expressed total RNAs, encompassing both coding and non-coding genes, in 162 esophageal cancer (EC) tissue samples compared to 11 normal control tissue samples, we analyzed RNAseq data from the TCGA database, as outlined in the Materials and methods section and illustrated though Flowchart in Figure 1. A total of 45 361 differentially expressed RNAs were identified in EC tissue samples compared to normal controls, which were re-annotated to Ensemble gene IDs using the “ensembldb” R package and categorized into various RNA types, including protein-coding genes (mRNAs), long non-coding RNAs (lncRNAs), miRNAs, pseudogenes, etc. Subsequently, applying a cutoff (log2FC > 1 + log2FC < −1 and FDR < 0.01), we extracted 3737 significantly differentially expressed RNAs, as shown in a volcano plot in Figure 2A. A total of 2222 genes were identified as significantly differentially expressed mRNAs (demRNAs), depicted in Figure 2B, with a corresponding heatmap of top-upregulated and top-downregulated demRNA panels shown on the left side of Figure 2B. Moreover, we obtained 966 significantly differentially expressed lncRNAs (delncRNAs), shown in the volcano plot in Figure 2C, with corresponding heatmaps of top-upregulated and top-downregulated delncRNAs panels shown on the left side of Figure 2C as heatmaps. The differently expressed mRNAs and lncRNA were sorted out using cut-off (log2FC > 1 + log2FC < −1 and FDR < 0.01). The overall above data suggest that good numbers of coding, as well as non-coding RNAs, are differently expressed in EC samples compared to healthy control samples. These candidate genes give us a lead to find the prognostic and immune-related lncRNAs in EC samples. Our study includes 81 patients with esophageal squamous cell carcinoma (ESCC) and 81 patients with esophageal adenocarcinoma (EAC) histological subtypes. We analyzed the data for EC subtype specificity but found no significant difference in the expression of lncRNAs. Hence, the data represents the whole 162 patient cohort.

Workflow of this study.

Identification of significant differentially expressed RNAs in Esophageal cancer. (A) Volcano plot showing differentially expressed total RNAs. (B) Volcano plot showing differentially expressed mRNAs and heatmap showing significantly upregulated and downregulated mRNAs. (C) Volcano plot showing differentially expressed lncRNAs and heat showing significantly upregulated and downregulated lncRNAs.
Establishment of immune-related mRNAs (IRmRNAs) and immune-related lncRNAs (IRlncRNAs) in EC
To discover differentially expressed immune-related long non-coding RNA in EC, we used the ImmPort database (https://www.immport.org/). Through the ImmPort database, we have found a total of 1793 immune-related genes that are associated with immune responses and immune system processes. Next, we overlapped these immune-related genes with 2222 differentially expressed mRNA identified from the previous. We found a total of 205 differentially expressed immune-related mRNA (deIRmRNA) in the EC patient’s sample, as illustrated through the Venn diagram in Figure 3A. Afterward, we performed Pearson correlation analysis using expression values of 205 deIRmRNA and 966 significant delncRNAs to find coexpressed immune-related lncRNAs in EC patients. As an output, we came up with 213 significantly correlated (P-value < .05) immune-related lncRNAs differentially expressed as illustrated through heatmap in Figure 3B using their respective correlation coefficients. Thus, we established a panel of 213 immune-related lncRNAs having a role in the immune response of EC patients.

Identification of Immune-related lncRNAs. (A) Venn diagram showing the overlapping between differentially expressed mRNAs (demRNAs) and immune genes from ImmPort database to find differentially expressed immune-related mRNA (deImRNA) in esophageal cancer. (B) Heatmap showing immune-related lncRNAs (IRlncRNAs) obtained from the correlation between delncRNAs and deIRmRNAs.
Twelve immune-related lncRNA signature predict prognosis of EC patients
We performed a Kaplan-Meier survival analysis on all 213 discovered immune-related lncRNAs to see which one has a stronger predictive prognostic value for EC. The EC patients were stratified into low and high-expression groups based on the median cut-off value for each IRlncRNA’s expression level. Kaplan-Meier and log-rank analyses were employed to evaluate the overall survival of these 2 expression groups. We used the R-package “ggplot2” for plotting Kaplan-Meier curves. Our analysis identified 12 IRlncRNAs with P-values less than .05 as significant prognostic lncRNAs. The identified 12 IRlncRNAs, namely are AC008687.2, AC134312.5, AL031587.3, AL353764.1, Heart and neural crest derivatives expressed transcript 2 antisense RNA 1 (HAND2-AS1), long intergenic non-protein coding RNA 261 (LINC00261), long intergenic non-protein coding RNA 1503 (LINC01503), long intergenic non-protein coding RNA 1614 (LINC01614), long intergenic non-protein coding RNA 2561 (LINC02561), myocardin-induced smooth muscle lncRNA Inducer of differentiation (MYOSLID), transmembrane 4L 6 family member 19 antisense RNA 1 (TM4SF19-AS1) and U62317.3 illustrated by Kaplan-Meier curves in Figure 4. Among these IRlncRNAs, AC134312.5 and HAND2-AS1 stands out as the most effective, with a P-value of .0034). These results underscore the potential of these identified 12 IRlncRNAs as promising prognostic markers in EC.

Kaplan-Meier curves to identify Immune-related lncRNAs having a significant role in esophageal cancer prognosis. (A) LINC01503, (B) LINC00261, (C) AL353764.1, (D) AL031587.3, (E) AC134312.5, (F) AC008687.2, (G) U62317.3, (H) TM4SF19-AS1, ()) MYOSLID, (J) LINC02561, (K) LINC01614, (L) LINC01503.
The expression of 12 prognostic IRlncRNAs has been found to have a notable and autonomous correlation with overall survival. In order to forecast the outcome of patients, these IRlncRNAs were amalgamated to create a signature. To determine a patient’s prognosis based on IRlncRNA expression, a risk score model was established using the coefficients derived from the Cox regression model along with the corresponding expression values of the IRlncRNAs. This model is depicted in the forest plot shown in Figure 5A. The formula for risk score calculation is ∑(regression coefficient × expression of lncRNA). According to this model, the 12 IRlncRNA prognostic risk score were computed for each EC patient. Figure 5B depicts the survival status of EC patients over the full cohort using a dot plot. All patients were classified into a high-risk group (n = 82) and a low-risk group (n = 80) according to the median risk score, as shown in Figure 5C. Subsequently, Kaplan-Meier survival analysis between low-risk and high-risk patients was performed. The result indicates that the low-risk patient’s survival rate was significantly higher compared to the high-risk group (P-value = .0049), as illustrated in Figure 5D. The 3-year survival rates in the high-risk and low-risk groups were 20.0% and 30.8%, respectively. The survival rate of the high-risk group was zero at 5-years, and that of the low-risk group was 10%. Thus, the Kaplan-Meier survival curve confirms that 12 IRlncRNA-based risk scores can significantly predict the overall survival between high-risk and low-risk groups. Moreover, we checked for association of risk score with various clinicopathologic features such as clinical stage, histologic grade, T-stage, N-stage and M stage, etc as shown in Figure 5E as heatmap. Results revealed significant association of risk score with histologic grade of EC patients (Supplemental Table S1).

Immune-related lncRNA signature-based risk score and its role in esophageal cancer prognosis. (A) Forest Plot showing results of Cox regression analysis on Immune-related lncRNA signature. (B) Dot plots showing the Survival status. (C) Risk score distribution of the entire EC cohort. (D) Kaplan-Meier curve showing the significant role of Immune-related lncRNA signature-based risk score in EC prognosis. (E) Heatmap showing the distribution of high and low risk patients with various clinicopathologic features such as clinical stage, histologic grade, T-stage, N-stage, and M stage, etc. Note: star sign show significant association.
Validation of the 12 immune-related lncRNA prognostic risk model
To assess the robustness and usability of the 12-identified immune-related long non-coding RNA (IRlncRNA) signature for EC prognosis, we performed additional validation by dividing the entire TCGA EC patients cohort into training (n = 81) and test groups (n = 81). As outlined in the Materials and methods section, the prognostic risk score for each patient was calculated based on the expression values of the 12 identified lncRNAs. In order to compare the sensitivity and specificity of risk score on the prognosis of EC patients, time-dependent receiver operating characteristics (ROC) analysis was performed on training, test, and whole cohorts. The area under the ROC curve (AUC) of the risk score in the training cohort is 0.67 (CI = 0.428 to 0.863), with a P-value of .002, as illustrated in Figure 6A. Meanwhile, the AUC of the risk score in the testing cohort is 0.812 (CI = 0.575 to 1.00) with a P-value of .007, as illustrated in Figure 6B. Moreover, the AUC of the risk score in the whole cohort is 0.712 (CI = 0.515 to 0.923) with a P-value of 0, as illustrated in Figure 6C. Thus, these AUC data indicate that 12 immune-related lncRNA prognostic signature is highly sensitive and specific to EC patient’s prognosis. Hence, these 12 immune-related lncRNA signatures can be explored as prognostic molecules for EC patients in the future.
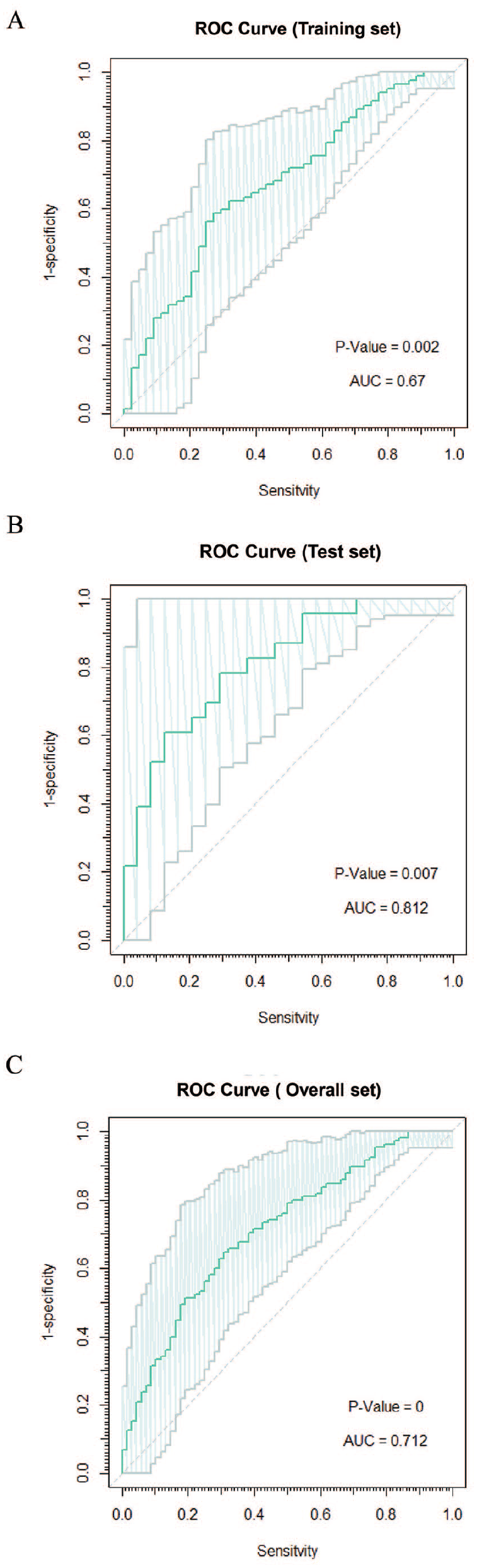
Validation of Immune-related lncRNA signature-based prognostic model. (A) The ROC curve for the training set with an AUC value of 0.67 shows a good prognostic value of the 12 Immune-related lncRNA-based prognostic signature. (B) The ROC curve for the test set with an AUC value of 0.81 shows a good prognostic value of the 12 Immune-related lncRNA-based prognostic signature. (C) The ROC curve for the entire set with an AUC value of 0.712 shows a good prognostic value of the 12 Immune-related lncRNA-based prognostic signature.
Functionally enriched biological role and pathways of 12 IRlncRNA
To confirm the downstream biological role of 12 IRlncRNAs in Immune functions, we performed GSEA for GO cellular components, molecular functions, biological processes, and KEGG pathways associated with 12 IRlncRNA signature. As a result, we obtained the top 10 enriched GO biological processes, which include many immune-related processes such as inflammatory response, response to cytokines, cytokines-mediated signaling pathway, and defense response, as shown in Figure 7A. Moreover, the top 10 significant GO molecular functions also involve some immune functions, such as chemokine activity, chemokine receptor binding, cytokine activity, and cytokine receptor binding as shown in Figure 7B. Furthermore, the top 10 GO cellular components involve vesicle lumen, secretory vesicles, secretory granules, external encapsulating structures, etc., which are well-known components of the cellular immune defense, as visualized in Figure 7C. Additionally, the top 10 KEGG pathways comprise natural killer cell-mediated cytotoxicity, toll-like receptor signaling pathways, cytokine-cytokine receptor interaction, JAK-STAT signaling pathways, chemokine signaling pathways, and, as illustrated in Figure 7D. These results indicate that our immune-related lncRNA signature has a significant role in many Immune-related processes and pathways.

Gene Set Enrichment Analysis for Gene Ontology (GO) and KEGG Pathways related to the IRlncRNA signature and its coexpressed genes. (A) The top 10 biological processes that have been greatly enriched. (B) The top 10 molecular processes that are significantly enriched. (C) The top 10 significantly impacted cellular activities. (D) The top 10 KEGG pathways that are significantly enriched. The length of the horizontal lines represents the gene numbers and FDR values in the index, which are represented by the colors in the index.
Effect of IRlncRNA signature and immune checkpoint blockade therapy
Previous investigation indicates that responsiveness to immune checkpoint blockade (ICB) treatment in individual patients may be correlated with expression of genes associated with immune checkpoint blockade therapy. 15 Thus, to investigate the potential involvement of IRlncRNA-based risk score in ICB immunotherapy in EC, we examined the difference in expression of ICB therapeutic targets (PD-1\PDCD1, PD-L1\CD274, TIM-3\HAVCR2, CTLA-4, PD-L2\PDCD1LG2, LAG3, TIGIT and VISTA\VSIR) in high and low-risk patients. The results showed that expression levels only PD-L1 (P-value = .048), PD-L2 (P-value = .002), and TIM-3 (P-value = .045) were significantly higher in low-risk patients compared to high-risk patients, as shown in Figure 8 via violin plot. This indicates that low-risk patients may have higher immunotherapeutic responses. Hence, our IRlncRNA-based risk score may play an important role in the intervention of ICB immunotherapy for patients with EC.
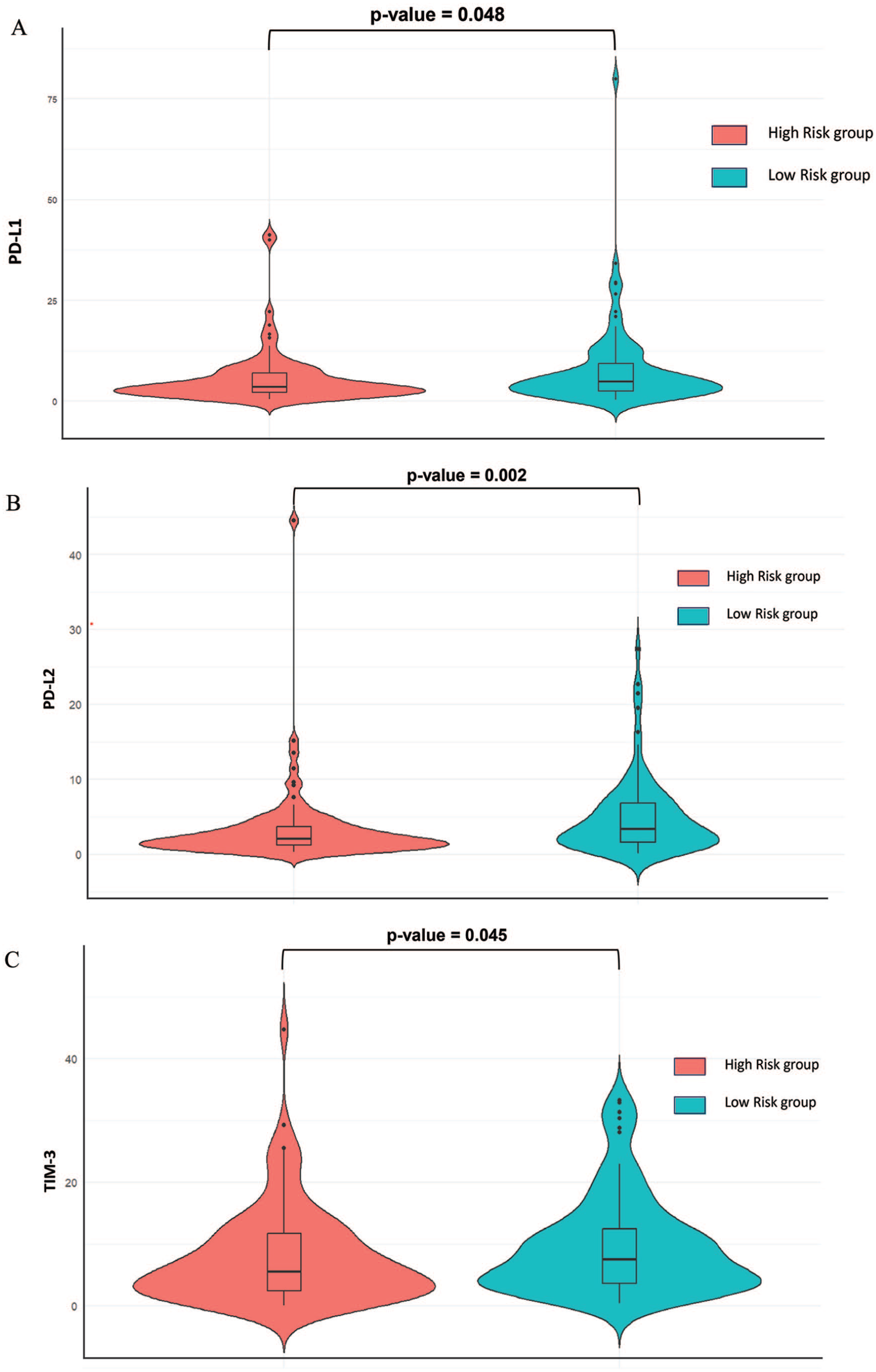
Association of Risk score and Immune checkpoints. (A) A violin plot shows a significant association between immune checkpoint PDL1 and risk score. (B) A violin plot shows a significant association between immune checkpoint PDL2 and risk score. (C) A violin plot shows a significant association between immune checkpoint TIM-1 and risk score.
Correlation between prognostic IRlncRNA signature based risk score and immune cells
To investigate the relationship between the immune-related lncRNA signature and antitumor immunity in patients with EC, we utilized the ESTIMATE algorithm to derive immune, stromal, and estimate scores for each individual. This approach enabled us to evaluate the tumor microenvironment (TME) in both high and low-risk groups. By comparing the stromal score (which reflects the presence of substrate cells in the tumor tissue), immune score (which indicates the infiltration of immune cells in the tumor tissue), and estimate score (a combined value of stromal and immune scores for each case), we were able to identify variations in infiltrating cells between the high-risk and low-risk groups. Notably, only the stromal scores were significantly elevated in the high-risk group (P-value < .00476), as depicted in Figure 9A. Additionally, we employed the CIBERSORT algorithm to analyze the immune cell infiltration landscape of individual EC patients. We examined the proportions of 22 different types of immune cells in both high and low-risk EC patients. Upon comparing the proportions of each immune cell between the 2 groups, we observed that plasma cells, resting CD4+ memory T cells, naive B cells, and regulatory T cells were significantly activated in the low-risk group compared to the high-risk group. Conversely, the fraction of M2 macrophages was notably higher in the high-risk group, as illustrated in the volcano plots presented in Figure 9B.

Immune status in high- and low-risk groups. (A) Comparison of immunological and stromal scores, as well as ESTIMATE scores in low- and high-risk groups. (B) Variation in the fraction of tumor-infiltrating immune cells between the 2 risk groups as per CIBERSORT algorithm.
Discussion
In recent times, various targeted immunotherapeutic approaches have been implemented for the treatment of cancer. These include immune checkpoint inhibitors such as PD-1, PD-L1/L2, and TIM-3, as well as adoptive tumor-infiltrating lymphocytes (TILs), cancer vaccines, and CAR T-cell treatments. 11 However, the prognosis and response to therapy can differ significantly among patients with EC. 30 One possible explanation for this variation is the heterogeneous nature of the disease, where each patient may elicit a unique immune response against cancer antigens. 31 Crucially, lncRNAs play a vital role in controlling the expression of genes associated with immune responses and activation. This leads to a diverse tumor immune microenvironment as it promotes the infiltration of various immune cells. 32 The significance of immune-related long non-coding RNA (IRlncRNA) signatures as prognostic markers has been established in multiple tumor types, including head and neck cancer, 33 Melanoma, 34 breast cancer, 35 and Glioblastoma. 36 Various studies have shed light on the potential role of lncRNAs in understanding and improving the treatment outcomes for EC patients.32,37 For example, lncRNAs such as MEG3 38 and LINC02096 (RIME) 39 show an immunosuppressive role in EC. Likewise, many significant lncRNA signatures were established in the recent past for predicting the immune landscape and prognosis for EC patients.40,41 However, few convincing prognostic markers exist to assist in identifying “high and low-risk” EC patients who may benefit from immunotherapy. In order to address this knowledge gap, we looked into the relationship between an immune-related long non-coding RNA signature that influences prognosis and its benefits for immunotherapy in patients with cancer. Given the widespread use of high-throughput technology and the ongoing development of data-sharing networks, the era of “big data” in tumor research has arrived. Exceptionally large amounts of multiple tumor data have been assembled in an internationally accessible database. 42 Previously, many researchers discovered chemokines-related, 40 stemness-related, 43 hypoxia-related, 44 Cuproptosis-related, 41 and ferroptosis-related 45 lncRNA signature having a role in EC prognosis using online databases. Similarly, Pang et al established the immune-related multi-lncRNA signature having a role in esophageal cancer prognosis using EC patient’s transcriptomic data from both TCGA and GEO databases. 27 Chen et al also added 5 key genes based ceRNA network having a role in the prognosis of TCGA EC patients. 46 However, in our study, we used an entirely different methodology, as shown in Figure 1, to identify and validate the 12 immune-associated lncRNA prognostic signature using transcriptome sequencing data of EC patients collected from only the TCGA database. Thus, in search for the best immune-related prognostic biomarkers for EC patients, we discovered 45 361 differentially expressed RNAs (deRNAs) in 162 esophageal cancer (EC) tissue samples compared to 11 normal control tissue samples. These RNAs were re-annotated to classify them as protein-coding genes (mRNAs) and long non-coding RNAs (lncRNAs), as illustrated and demonstrated in Figure 2, respectively. Top Upregulated mRNAs include ATP4B, ATP4A, AQP4, PGA5, GKN1-2, and ETNPPL. Among these gastric H+/K+ ATPase proton pump (ATP4A/ATP4B) and pepsinogen (PGA5) are known to play role in carcinogenesis of Barrett’s esophagus (BE) associated EAC. 47 Gastrokines (GKN1-2) plays role in gastric cancer progression. Moreover, Ethanolamine-phosphate phospho-lyase (ETNPPL) contributes to the diagnosis, prognosis, and therapy of hepatocellular carcinoma. 48 Top downregulated mRNAs include MAGEA1-6-11, PAGE2B, GPR50, PAEP, KRTAP4-1, KRT81, ACTL8, CSAG2. Among these MAGEA family proteins have been established as prognostic as well as diagnostic marker for ESCC and EAC both with immunotherapeutic efficacy.49 -51 Whereas, P Antigen Family, Member 2B (PAGE2B) was established as one of the 13 immune related genes having role in prognosis of clear cell renal cell carcinoma. 52 Next, G Protein-Coupled Receptor 50 (GPR50) has been found differentially expressed in various cancer such as breast cancer, 53 hepatocellular carcinoma, 54 kidney renal papillary cell carcinoma, 55 and glioma. 56 Progestagen associated endometrial protein (PAEP) is known for its role as immune system modulator in reproduction but recently its role in various cancer has been reported such as NSCLC, 57 melanoma, 58 endometrial cancer, 59 etc. Keratin 81 (KRT81) and keratin associated proteins 4-1 (KRTAP4-1) role as epithelial cell marker is well established and role in cancer targeted drug delivery is underway. 60 Actin-Like Protein 8 (ACTL8) is also studied for its potential as prognostic biomarker for various cancer such as colorectal cancer, 61 lung cancer, 62 glioma, 63 head and neck cancer, 64 TNBC 65 etc. Likewise, chondrosarcoma-associated gene 2 (CSAG2) has also been thoroughly studied for its role as biomarker for various cancer such as ovarian cancer, 66 osteosarcoma, 67 and chondrosarcoma. 68
Furthermore, to find the immune genes having a significant role in immune response and function, we used the immune genes dataset from the ImmPort database as a reference and obtained 205 differentially expressed immune-related mRNA (deIRmRNA) in EC samples. Since lncRNAs coexpressed with Immune genes share similar biological functions, 69 we employed Pearson correlation analysis and obtained 213 immune-related lncRNAs (IRlncRNAs) that were highly coexpressed with 205 deIRmRNA, as shown in Figure 3. Finally, to find the lncRNAs that can independently predict the prognosis of EC patients, Kaplan-Meier survival analysis was employed resulting in 12 IRlncRNAs named AC008687.2, AC134312.5, AL031587.3, AL353764.1, HAND2-AS1, LINC00261, LINC01503, LINC01614, LINC02561, MYOSLID, TM4SF19-AS1, and U62317.3 as displayed in Figure 4. Surprisingly, we also obtained LINC01614 as a common lncRNA with the previous signature. 27 The reason for the difference in previous signature could be due to distinct methodology or genetic heterogeneity in individual patients. Till now, the expression of only LINC00261, 70 LINC01614, 71 LINC01503, 72 and HAND2-AS1 73 was validated and found upregulated in esophageal cancer tissues and cells. Thus, to improve the predictive accuracy of 12 IRlncRNAs, we combined them and developed 12 IRlncRNA-based risk scores using the formula mentioned in section 2.5. Further, the Kaplan-Meier survival curve in Figure 5 confirmed that 12 IRlncRNA-based risk scores can significantly predict overall survival between high-risk and low-risk patients.
Interestingly, we also validated the robustness and utility of the 12-IRlncRNA signature for EC prognosis using time-dependent receiver operating characteristics (ROC) analysis on training, testing, and entire cohorts as a part of internal validation, as shown in Figure 6. The AUC data show that the 12 IRlncRNA prognostic signature is highly sensitive and specific to EC patient prognosis, implying that the 12 IRlncRNA signature can be studied as a prognostic model for EC patients in the future. In order to confirm the biological role of 12 IRlncRNAs in immune functions, GSEA was performed for GO biological processes, molecular functions, cellular components, and KEGG pathways associated with their coexpressed genes, as illustrated in Figure 7. The results confirmed the active role of 12 IRlncRNA in immune-related processes like inflammatory, cytokine, and defense responses, along with other cytokine and chemokine activities. Also, some enriched biological functions such as cell motility, locomotion, taxis, and response to lipids also show role in cancer aggressiveness and metastasis. 74 Moreover, KEGG pathways associated with 12 IRlncRNA signature include many pathways associated with cancer like TGF beta signaling and JAK/STAT signaling pathway also having role in activation of interleukins, cytokines, and growth factors. 75 Moreover, immune specific signaling such as toll-like receptor signaling pathways and natural killer cell-mediated cytotoxicity chemokine signaling pathways are also highly enriched having important roles in tumor microenvironment. Furthermore, using the ESTIMATE algorithm, we compared immune, stromal, and estimate scores to evaluate the tumor microenvironment and found that stromal scores were significantly higher in the high-risk group (Figure 9). The percentage of stromal cells in TME indicates the stromal score, and stromal cells are one of the most important components of TME. 76 Tumor stroma, particularly cell components, plays an important role in the development of EC. Thus, explaining the higher percentage of stromal cells in the high-risk group. Additionally, the CIBERSORT algorithm revealed that plasma cells, naive B cells, resting CD4+ memory T cells, and regulatory T cells were activated in the low-risk group. The results were in harmony with previous studies that confirmed that high-risk cancer patients have suppressed immune response compared to low-risk cancer patients. 77 In contrary, tumor infiltrating M2 macrophages were found significantly higher in the high-risk group.
We also explored the role of the IRlncRNA-based risk score in immune checkpoint blockade (ICB) immunotherapy in EC patients. It was found that PD-L1 (P-value = .048), PD-L2 (P-value = .002), and TIM-3 (P-value = .045) expression levels were significantly higher in low-risk patients compared to high-risk patients (Figure 8). Suggesting that low-risk patients may have higher immunotherapeutic responses and giving crucial insights for ICB immunotherapy intervention. Although these findings confirm the role of 12 IRlncRNAs signature in prognosis and immunotherapeutic response in EC patients using a bioinformatics approach. However, the validation and functional characterization of these IRlncRNAs should be done using in-vitro cell-based molecular biology experiments such as qRT-PCR.
Conclusion
In this study, we have successfully identified and validated a set of 12 IRlncRNAs signature that hold promising potential for assessing the risk and prognosis of patients with EC. Through the analysis of survival data and the utilization of ROC curves, we have unequivocally demonstrated the strong predictive capabilities of our signature in determining the survival outcomes of EC patients. The findings of our study not only hold immense significance in prognostic prediction but also offer valuable insights for guiding future immunotherapy approaches in individuals diagnosed with esophageal cancer.
Supplemental Material
sj-docx-1-cix-10.1177_11769351241276757 – Supplemental material for Immune-Related Long Non-Coding RNA Signature Determines Prognosis and Immunotherapeutic Coherence in Esophageal Cancer
Supplemental material, sj-docx-1-cix-10.1177_11769351241276757 for Immune-Related Long Non-Coding RNA Signature Determines Prognosis and Immunotherapeutic Coherence in Esophageal Cancer by Vivek Uttam, Harmanpreet Singh Kapoor, Manjit Kaur Rana, Ritu Yadav, Hridayesh Prakash, Manju Jain, Hardeep Singh Tuli and Aklank Jain in Cancer Informatics
Footnotes
Acknowledgements
The Central University of Punjab, Department of Zoology, supported the research by providing adequate research facilities and funds to Prof. Aklank Jain. University Grants Commission (U.G.C.), New Delhi, supported in the form of Junior Research Fellowship (J.R.F.; U.G.C. ref. no.; 191620043900) under the NFSC scheme to Vivek Uttam.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration Of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
A.J. conceived the original idea; V.U. and H.S.K. planned the framework and performed the data analysis; V.U. and R.Y. wrote the manuscript, V.U. prepared the figures, and formatted the final manuscript according to journal guidelines; V.U., M.K.R., H.P., M.J., H.S.T., and A.J. contributed to the final editing of the manuscript; A.J. supervised and supported the research.
Availability of Data and Materials
The analyzed raw data is publicly available at The Cancer Genome Atlas (TCGA) database of the United States National Cancer Institute.
Ethics Approval and Consent to Participate
As the data is publicly available at The Cancer Genome Atlas (TCGA) database Ethical approval and consent of participation are not applicable.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.