
Abstract
Genome instability of cancer cells translates into increased entropy and lower information processing capacity, leading to metabolic reprograming toward higher energy states, presumed to be aligned with a cancer growth imperative. Dubbed as the cell adaptive fitness, the proposition postulates that the coupling between cell signaling and metabolism constrains cancer evolutionary dynamics along trajectories privileged by the maintenance of metabolic sufficiency for survival. In particular, the conjecture postulates that clonal expansion becomes restricted when genetic alterations induce a sufficiently high level of disorder, that is, high entropy, in the regulatory signaling network, abrogating as a result the ability of cancer cells to successfully replicate, leading to a stage of clonal stagnation. The proposition is analyzed in the context of an in-silico model of tumor evolutionary dynamics to illustrate how cell-inherent adaptive fitness may predictably constrain clonal evolution of tumors, which would have significant implications for the design of adaptive cancer therapies.
Keywords
Introduction
The evolving genetic diversity and phenotypic plasticity of cancer cells make cancer a moving target for therapeutic interventions. Despite the increasing efficacy of available cancer therapies, resistant cancer cells persist after remission as a minimal residual disease which would ultimately lead to recurrence, therapeutic resistance and metastasis. Constraints on cancer evolutionary dynamics, such as the incidence order of genetic alteration events, may point to vulnerabilities that can be targeted to achieve effective management or cure.
Cancer is a nonlinear dynamical system driven by stochastic genetic events involving driver and passenger somatic mutations, 1 copy number alterations, 2 and structural variations, including translocations, chromosomal loss or gain and whole genome doubling. 3 A recent high accuracy DNA duplex sequencing of colorectal tumors shows that every DNA base is mutated in at least one cancer cell. 4 The assertion of such hyper genotypic diversity at diagnosis highlights the near boundless potential of tumor cells for phenotypic transformations and the vast space of possible trajectories of survival and proliferation they could explore under environmental and therapeutic selection pressures. Herein lies the limitations of therapeutic interventions that do not integrate the evolutionary perspective of cancer, first proposed by Nowell 5 and increasingly recognized as the most fitting viewpoint in the search for effective cancer treatments. 6 In particular, cancer evolutionary dynamics impose an intrinsic barrier to the cure or effective management of cancer using the common therapeutic paradigm of killing as many cancer cells as possible, argued to be evolutionary unsound. 7 Indeed, traditional therapeutic strategies, which are often based on genotoxic agents, eradicate therapy-sensitive cancer cells, leaving therapy-resistant cells to subsequently fuel resistance and disease recurrence, 8 as the most critical challenges in the fight against cancer. Treatment strategies that are designed to anticipate the evolutionary dynamics of tumor progression and accordingly preempt the adaptive survival of cancer cells are expected, in principle, to yield improved patient outcomes. However, this will hinge on the feasibility of predicting tumor progression trajectories. Genome wide studies have revealed genotypic patterns underlying cancer progression, whereby different cancer stages involve different sets of driver mutations and copy number alterations.9-11 Although uncovering genotypic patterns in tumors’ histories may inform the design of therapeutic strategies, predicting the course of tumor evolution would require delineating the dynamics of cancer progression as they relate to what evolution acts on, that is, the phenotype. Furthermore, while stochastic genetic events shape the emergence of distinct metabolic and cell proliferation phenotypes, the evolutionary dynamics of tumor progression are ultimately determined by selection pressures of the tumor microenvironment (TME) and therapeutic interventions acting on the phenotypic diversity of cancer cells. However, this does not preclude the existence of cell-intrinsic mechanisms that shape cancer cell fitness and modulate selection pressures. Indeed, whereas cancer evolutionary drivers are stochastic, the nonlinear tumor response to therapy is predictably adaptive to therapeutic challenges and fosters therapeutic resistance. The cell signaling and metabolic networks are the integrators of genetic alterations into observable phenotypes and hence constitute an appropriate level of abstraction to reason about potential mechanisms underlying any cell-intrinsic fitness adaptation modulating evolutionary selection. The adaptive fitness of cancer cells is ultimately manifested through the emergence of trajectories of confluence between metabolic reprograming and cell fate choices that support clonal proliferation.
The proposed necessity of concordance between cell fate decision-making and altered metabolism to support cancer proliferative growth is rather intuitive given the feedback loops that couple the cell signaling and metabolic networks12,13. In other words, the set of potential cell fate choices of cancer cells may be limited by the integrated effects of genetic alterations on oncogenic signaling in closed loop with altered metabolism. The limitation on cancer cell fate choices, would ultimately translates into evolutionary constraints on tumor progression. That is, the signaling-metabolism feedback loop dampens the stochastic effect of genetic alterations, limiting as a result the set of possible cell fate trajectories that would be available to drive tumor proliferative growth. Characterizing the corresponding constraint as a function of genetic alterations may provide an avenue for one-step ahead predictions of evolutionary dynamics, which would be instrumental to the rational development of adaptive cancer therapies.
Phenotypic Measure of Genetic Alterations
Recent studies of cancer evolutionary history have shed light on the sequence of genetic events implicated in different cancer types.9,11 Although trends and patterns of cancer evolution may be used to stratify patients for treatments, the semblance of order represented by such patterns is still wrapped in a stochastically driven evolution whose prediction would require the definition of measures that go beyond the stochasticity of the genotypic drivers and focus rather on the phenotypic diversity on which evolution acts. The generation of such phenotypic diversity is necessarily bound to the pathway-integrated effects of genetic alterations and to the dynamics of the closed loop system that couples the signaling and metabolic networks of cancer cells (see Figure 1) and lead to the reprograming of metabolism, cell death evasion, and sustained proliferation. 14

Cell cycle regulation. The cell cycle is under the regulatory control of the signaling network and is dependent for its progression on the output of the metabolic network. On the other hand, the signaling and metabolic networks are coupled through feedback signals, making the effect of genetic alterations reverberates through the entire signaling-metabolic system. The resulting perturbations of the coupling between signaling and bioenergetics would expectedly be a determinant of the growth and proliferation dynamics of cancer.
The intertwined metabolic and cell fate decision-making pathways (eg, cell division, differentiation, senescence and apoptosis) constitute clearly delineable dimensions along which the effects of genetic alterations are integrated to yield the survival and proliferation potentials of cancer cells, which ultimately determine their evolutionary fitness. This highlights the need to quantify the dysregulation of signaling and metabolic pathways in order to characterize the phenotypic trends of cancer evolution. Network entropy, defined based on Kolmogorov–Sinai entropy,
15
has been applied to the study of various aspects of protein-protein interaction networks, including their robustness and the evolution of their structures.
16
Network entropy was also used to discriminate between cancer and normal cells
17
and predict sensitivity to cancer drugs.
18
Given the dominance of various set of oncogenic pathways in the oncogenesis and progression stages of different cancers, the notion of pathway entropy is introduced with the explicit consideration of oncogenic pathways that are relevant to the cancer at hand. It is used as a measure of the rewiring of oncogenic signaling pathways which drives clonal exploration of proliferation trajectories that feed tumor growth. The cell signaling network is modeled as a directed graph where nodes represent signaling proteins, while directed edges represent protein-protein interactions. Pathway entropy

Given

Pathway entropy was shown to discriminate between cancer and normal cells for lung squamous cell carcinoma (LUSC) and colorectal adenocarcinoma (COADREAD). 19 It can be computed for any set of signaling pathways as a measure of the effect of genetic alterations on the proliferative potential and therapeutic resistance of cancer cells. Depending on the cancer type under consideration, pathway entropy may be used as a resistance biomarker by focusing on the most relevant oncogenic pathways, such as the consideration of the MET, ALK, and EGFR pathways in the treatment for NSCLC (Non-Small Cell Carcinoma) using tyrosine kinase inhibitors. Ultimately, pathway entropy is a phenotypic measure of genetic alterations, formulated at the pathway level of abstraction considered for reasoning about the hallmarks and treatments of cancer and is hence plausible as a source of insight into cancer evolutionary dynamics.
Cell Adaptive Fitness
The entropy of cell signaling pathways provides a quantification of the genetically-induced perturbations to cell fate decision-making and bioenergetics in cancer cells. The regulation of the cell life-cycle, cell fate decision-making, and metabolic activities depend on the processing of signals and cues from the environment. The underlying cell information processing capacity is represented by the cell information state, denoted
Cells are nonlinear, stochastic dynamical systems that can be described using the trajectories of their information and energy states. The cumulative increase of genetic alterations in cancer cells perturb signaling regulation and lead to a rise in signaling entropy and a higher instability of the metabolic output. On the other hand, an increase in signaling entropy induces a weakening of the cell information processing capacity, leading to a higher irregularity of cancer cell-fate decisions compared to normal cells. The corresponding stochasticity of information and energy states enables transformed cells to sample a large space of phenotypes and adapt their survival trajectories under the selective pressure of temporally and spatially changing nutritional and stimulatory conditions of the tumor microenvironment. Such cell-level stochasticity underlies the emergence of population-level (i.e., tumor) evolutionary dynamics, including therapeutic resistance. Evolutionary models of tumor progression often assume driver mutations to confer a fixed average selective advantage. 21 However, any regulatory constraints imposed by the feedback loops between signaling and metabolism on cell-fate decision-making and metabolic states will translate into constraints on the set of viable clonal growth and proliferation trajectories. This would modulate the selective advantage conferred by genetic alteration events, including passenger mutations, whose contributions are also implicated in tumorigenesis. 1 In particular, we propose that the increase of signaling entropy due to genetic alterations will lead to a decline of the information processing capacity of cancer cells and an increase of their bioenergetic capacity, which will eventually peak and begin declining when the cell information processing capacity becomes insufficient to support the normal regulation of cellular processes. This proposition, that we call the cell adaptive fitness, is an expression of a presumed imperative of growth that is hard wired in the genetic and proteomic circuitry of living organisms, whereby cellular bioenergetic processes evolve to transform all available nutrients unless otherwise regulated by the signaling network. In other words, cells optimize their energy production and consumption for metabolic sufficiency when their life-cycle processes are regulated, which corresponds to high cell information states. In contrast, cancer cells have varying degrees of dysregulated signaling pathways and obstructed flows of regulatory information that translate into relaxed metabolic regulation. This permits cancer cells to indulge in siphoning more nutritional resources through runway metabolic activities, achieving as a result higher metabolic states to support fast growth and proliferation. However, the inverse variation between cell information and energy states is expected to break down at lower information states. Indeed, as the entropy of the signaling network increases with the increasing burden of genetic alterations, the information processing capacity of corresponding clones and sub-clones will eventually dip below a putative minimum threshold necessary to achieve a homeostatic regulation of the cell life-cycle. Below such putative threshold, cell fate decision-making capability would become too compromised to sustain a stable growth and proliferation program. By the same token, the signaling-regulated enzymatic co-cooperativity of the metabolic network would also break down and would no longer be able to yield the high metabolic output needed by fast proliferating cancer cells. This transition point ushers the convergence of cancer cells toward lower energy and information states (see Figure 2), characterized by high genomic instability where successful replications would be rare. 22

Cell information and metabolic/energy states as functions of the entropy of signaling pathways. In cancer cells, the increase of signaling entropy, driven by the accumulation of driver mutations, corresponds to a decline in the information processing capacity underling cell fate decision-making and metabolic regulation. This decline translates into relaxed control over metabolic activities leading to the reprograming of metabolism with a corresponding high metabolic output characterizing the proliferative phenotype of cancer cells.
Although direct measurements of cell energy and information states may not be currently feasible, the profiles of their trajectories as functions of signaling entropy provide an insight into the multi-stage clonal evolution shaped by accumulating genetic alterations. The proposed relationships between entropy, energy and information (see Figure 2) may be partially corroborated by reconstructed cancer mutational histories through whole-genome sequencing analysis. 9 For instance, only 9 genes are affected by 50% of early clonal driver mutations compared to 35 genes for 50% of late clonal driver mutations, 9 indicating that the arrow of tumor evolution corresponds to higher numbers of genes and signaling pathways being affected, driving as a result the increase of cell signaling entropy, and eventually affecting the viability of cancer cells 23 . In addition, mutational signatures such as defective mismatch repair exhibit a directed temporal increase from clonal to sub-clonal evolutionary stages of tumor progression. 9 Furthermore, while the increased diversity of genes affected in the late stages of tumor progression may be a source of clonal adaptation, high genomic instability compromises the viability of cancer cells. Taken together, these observations inspired the proposed phenomenological relationships between bioenergetics, signaling entropy, and information processing as an explanation of the potential mechanisms linking the accumulation of genetic alterations to the dynamics of clonal evolution.
Evolutionary Dynamics of Cancer
The nonlinear relationship between cell information and energy states is explored as a potential evolutionary constraint that modulates the selective advantage conferred to cancer cells by acquired genetic alterations. More specifically, clonal evolution is proposed to be characterized by a multi-stage progression trajectory where each stage is delineable by phenotypes that are determined by the dynamics of coupling between cell-fate decision making and metabolism. As cancer cell entropy increases with the number of driver mutations, the coupling between cell bioenergetics and cell-fate decision-making enforces an upper limit on clonal potential for expansion. While accumulating driver mutations are permissive of intensified bioenergetic activities, they are also the source of high genetic instability that leads to metabolic insufficiency and replication failures. This proposition is explored using an extension of the tumor progression model of Bozic et al.
21
The model is built on a Galton-Watson branching process,
24
starting with a single cell that either divides or takes an alternate fate of “stagnation,” which may be differentiation, senescence or death.
21
The model tracks the number of cells having

Here,



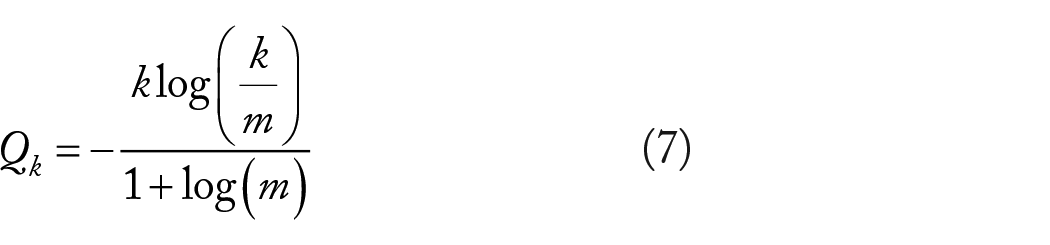
Here
Simulating the tumor evolutionary model using
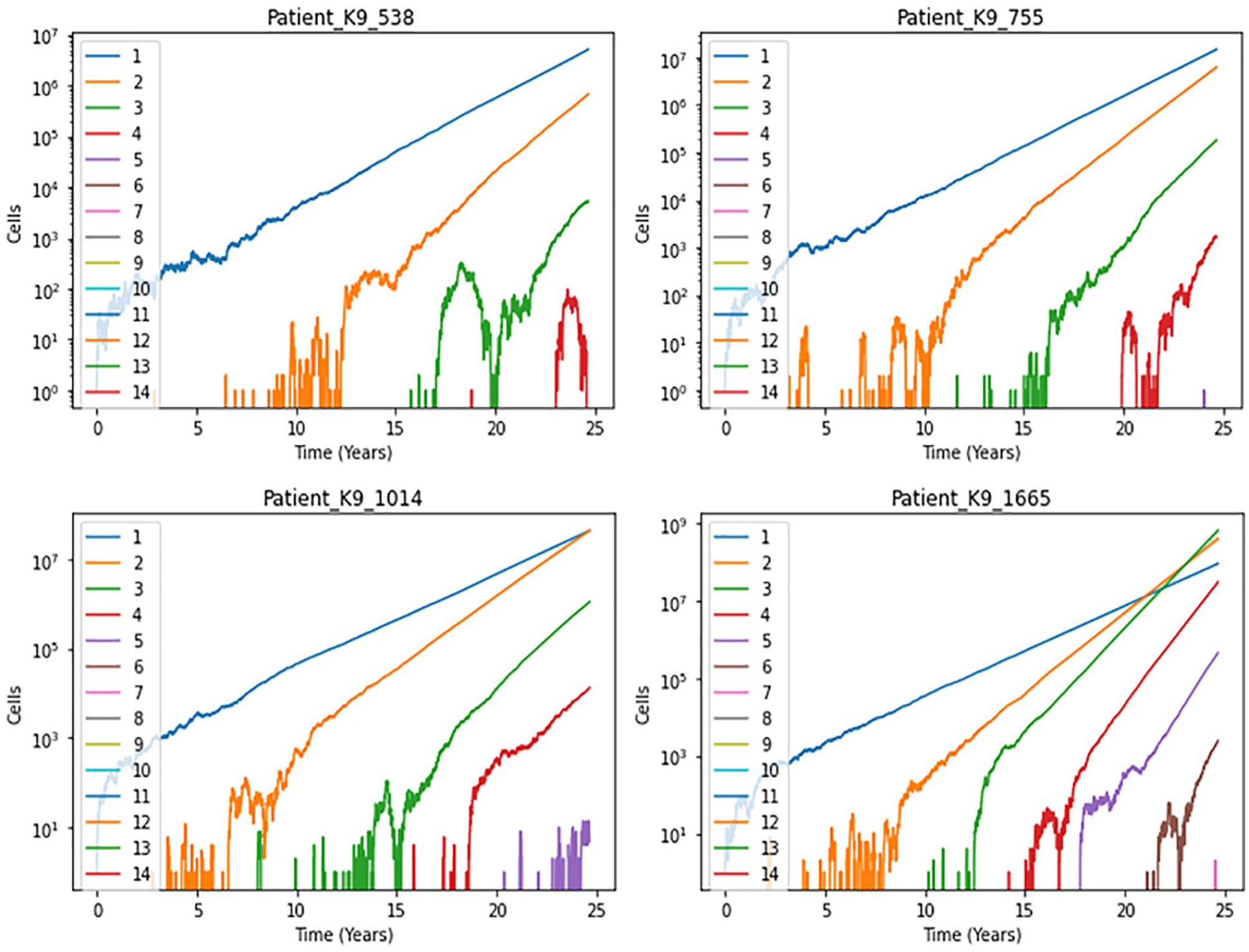
Constrained evolutionary dynamics of tumor progression. Clone size (number of cells with a given number of driver mutations) versus the age of the tumor. Four simulation runs of the extended tumor progression model with a clone-dependent selective advantage. The widely varying dynamics of tumor growth is distinguished by a marked constraint on the number of emerging clones. Simulation runs are labeled as hypothetical patients.
The simulation illustrates the working of the conjectured cell-inherent adaptive fitness as a potential source of constraints on evolutionary dynamics of tumor progression. In particular, the overwhelming majority of simulation runs did not yield any tumors, highlighting the challenge of predicting cancer occurrence based on genomic profiling. The model recapitulates the variability of tumor progression trajectories and points to a constrained clonal expansion. For instance, the final tumor size for the overwhelming majority of patients rarely reaches the carrying capacity

Tumor average entropy. (a) 11 patients, (b) 14 patients, (c) 24 patients, and (d) 24 patients. Four simulation runs (a–d) of the stochastic tumor progression model yielding different numbers of tumors, labeled as hypothetical patients. Each color represents the tumor average entropy as a function of time for one hypothetical patient. Patients are differentiated by starkly distinct trends of the tumor average entropy.
Implications for Therapeutic Strategies
The proposed perspective on tumor evolutionary dynamics has the potential to support the design of therapeutic strategies that are based on eco-evolutionary principles such as adaptive therapies. 8 Adaptive combination therapies are promising approaches for the management of drug resistance and residual disease. Their effectiveness hinges on the accurate monitoring of tumor burden and requires, at a minimum, a one-step ahead prediction of clonal composition. Such monitoring enables the consideration of the disease’s evolutionary potential which leverages genetic diversity and epigenetic plasticity to sustain robust trajectories of growth under therapeutic targeting.6,29-31 Indeed, tumors with highly diverse clonal compositions are more likely to harbor malignant cell phenotypes,32,33 and are associated with worst outcomes for various cancers.34-39 Estimating cancer evolutionary dynamics is therefore necessary for the development of effective therapies, including adaptive therapies aiming to control cancer based on ecological principles of clonal competition.40,41 Although the nonlinear dynamics of cancer and the uncertainty on the knowledge of the initial conditions of carcinogenesis limit the predictability of cancer dynamics,42-45 the postulated existence of a cell-inherent adaptive fitness may enable the estimation of clonal dynamics up to a time horizon that may be sufficient to support effective adaptive therapies. Monitoring tumor clonal and sub-clonal compositions using ctDNA liquid biopsy 46 would provide the mean to estimate clonal and tumor average entropies. Given the multi-stage clonal evolution, conjectured to be marked by a monotonically increasing clonal entropy, a one-step-ahead prediction of stage-transitions could serve as a feedback for the adaptation of cancer therapy. In this context, multiple drugs may be combined sequentially, concurrently or using a mixed schedule thereof to control the disease and prevent the emergence of therapeutic resistance. Monitoring the patterns of expansion-stagnation of specific clones, would inform precision therapeutic targeting that needs to be applied at each round of therapy. This may involve continuing with the same drugs, switching between drugs used in previous rounds of therapy or introducing new drugs, based on the observed evolution stages of tumor clones. For example, when a clone is predicted to enter a stagnation stage based on their estimated signaling entropy and energy-information profile, targeting a driver mutation using TKIs (tyrosine kinase inhibitors) would increase toxicity without significant therapeutic benefits. On the other hand, targeting clones before an anticipated commencement of expansion would have a more significant effect on outcome. However, given the evolutionary dynamics of tumor progression, every therapeutic action will have an adaptive tumor survival reaction. Therein lies the potential utility of the proposed evolutionary model as a predictive component of clinical decision-support systems that may be conceived to assist oncologists in the design of adaptive cancer treatment strategies that leverage the advantage of anticipating cancer evolution toward thwarting therapeutic resistance.
The prediction of clonal, evolutionary stage transitions would also be of utility to adjuvant therapies, which are designed to eliminate small residual populations of cancer cells associated with minimal residual disease (MRD). For instance, colorectal cancer (CRC) cells were shown to have an equal potential to become drug-tolerant persister (DTP) in response to therapy, 47 and that such transitions to the DTP state is fostered by an increase of mutation rate. 48 In light of the proposed conjecture, the timing of clonal expansion would coincide with an increase of clonal signaling entropy induced by heightened mutability and a corresponding switch to a proliferative state. Given this observation, longitudinal MRD monitoring, using ctDNA liquid biopsies, could include the tracking of clonal signaling entropy to distinguish between high entropy clones converging toward senescence from the low entropy clones having a higher potential for drug resistance. This would inform the design of more effective adjuvant therapies that focus on the latter population of residual cancer cells (i.e., having lower entropy) to preempt the emergence of DTP clones. Such therapeutic approach, which may be qualified as a precision adjuvant therapy, would be an improvement on current adjuvant therapies used for various cancers to prevent recurrence and metastasis.
Discussion
The proposed cell adaptive fitness conjecture implies that clonal architecture is characterized by an increasing average entropy and is shaped by a selective advantage
The conjecture is an attempt to explain evolutionary dynamics of tumor growth through the lens of the coupling between signaling and metabolic pathways as the mediator of dynamic interactions between the tumor and its co-evolving heterogenous microenvironment. It is also an attempt to offer analytic insight into the causal chain linking pathways as targets of therapeutic actions, and tumor growth dynamics as indicators of clinical outcome. Full evidentiary support for the conjecture will ultimately rest on future clinical validations of its characterization of clonal evolution and the extent to which such characterization is successful in enabling improvements of cancer treatment strategies. In this respect, longitudinal monitoring of tumor mutational changes using liquid biopsies and next-generation sequencing (NGS) may be used in future efforts to build clinical support for the proposed conjecture and validate the corresponding model of tumor evolutionary dynamics. Data obtained through longitudinal tumor monitoring may be used in conjunction with known patterns of cancer evolutionary histories 9 and key oncogenic pathways to track clonal architecture and corresponding clonal and tumor entropies. Conjecture-supported tumor evolutionary trajectories would then be analyzed in relation to disease progression states, to be asserted using currently applicable protocols, as a first step toward developing supporting evidence for the clinical validation of the proposed conjecture.
Conclusions
The cell adaptive fitness conjecture embodies, to the author’s knowledge, the first ever proposition put forth about the existence of an inherent cell adaptation mechanism wired in the coupling between cell signaling and metabolism to maintain the cell potential for survival, growth and replication. Although driver mutations underly oncogenesis and tumor evolutionary dynamics, their accumulation will ultimately engender the emergence of evolutionary constraints driven by the coupling between signaling and metabolism, and manifested as a cell adaptive fitness that confers a varying clonal selective advantage and leads to phenotypic convergence. One of the consequences of cell adaptive fitness is that the increasing entropy shapes the phenotypic landscape of clonal evolution with clear implications for therapy. Low-entropy early clone variants are likely to harbor stem-like therapy resistant tumor cells, while high-entropy late clone variants are overwhelmed with senescence. The focus of cancer treatments on the proliferating phenotype, that is, having an entropy that maximizes the proliferation potential, should therefore be complemented with resistance management strategies that target the stem-like, low-entropy early clone variants.
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work and its publication were supported by Toronto Metropolitan University.
Declaration Of Conflicting Interests:
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Not applicable.