
Abstract
Background:
Colorectal cancer is the third largest cause of cancer-related mortality worldwide. Although current treatments with chemotherapeutics have allowed for management of colorectal cancer, additional novel treatments are essential. Intervening with the metabolic reprogramming observed in cancers called “Warburg Effect,” is one of the novel strategies considered to combat cancers. In the metabolic reprogramming pathway, pyruvate dehydrogenase kinase (PDK1) plays a pivotal role. Identification and characterization of a PDK1 inhibitor is of paramount importance. Further, for efficacious treatment of colorectal cancers, combinatorial regimens are essential. To this end, we opted to identify a PDK1 inhibitor using computational structure-based drug design FINDSITEcomb and perform combinatorial studies with 5-FU for efficacious treatment of colorectal cancers.
Methods:
Using computational structure-based drug design FINDSITEcomb, stearic acid (SA) was identified as a possible PDK1 inhibitor. Elucidation of the mechanism of action of SA was performed using flow cytometry, clonogenic assays.
Results:
When the growth inhibitory potential of SA was tested on colorectal adenocarcinoma (DLD-1) cells, a 50% inhibitory concentration (IC50) of 60 µM was recorded. Moreover, SA inhibited the proliferation potential of DLD-1 cells as shown by the clonogenic assay and there was a sustained response even after withdrawal of the compound. Elucidation of the mechanism of action revealed, that the inhibitory effect of SA was through the programmed cell death pathway. There was increase in the number of apoptotic and multicaspase positive cells. SA also impacted the levels of the cell survival protein Bcl-2. With the aim of achieving improved treatment for colorectal cancer, we opted to combine 5-fluorouracil (5-FU), the currently used drug in the clinic, with SA. Combining SA with 5-FU, revealed a synergistic effect in which the IC50 of 5-FU decreased from 25 to 6 µM upon combination with 60 µM SA. Further, SA did not inhibit non-tumorigenic NIH-3T3 proliferation.
Conclusions:
We envision that this significant decrease in the IC50 of 5-FU could translate into less side effects of 5-FU and increase the efficacy of the treatment due to the multifaceted action of SA. The data generated from the current studies on the inhibition of colorectal adenocarcinoma by SA discovered by the use of the computational program as well as synergistic action with 5-FU should open up novel therapeutic options for the management of colorectal adenocarcinomas.
Keywords
Background
Colon cancer ranks third among all cancers with regard to incidence as well as mortality worldwide. According to cancer statistics, over 140 000 people died of colorectal cancer in 2016 1 and >104 000 new cases of colon and >43 000 new cases of rectal cancers were reported in 2020. In the early stages of colon as well as rectal cancer, resection could be helpful, accompanied by neoadjuvant chemotherapy, but 22% of stage II and III cancer patients exhibit relapse of the cancer. 2 Further, administration of chemotherapeutic drug like 5-FU is the preferred choice of treatment along with leucovorin, and oxaliplatin 3 ; but many of these cancers exhibit resistance to 5-FU. 4 Therefore, to manage colorectal cancer, better therapeutic strategies are essential. Combination of novel entities affecting key signaling pathways along with 5-FU could be one of the novel strategies to be considered. Cancer cells’ uniqueness when compared to normal cells is their energy requirement. To meet this requirement, metabolic reprogramming occurs in these cells. This metabolic reprogramming called the Warburg Effect, was discovered by Otto Warburg in 1924. This effect is characterized by reliance on glycolysis for energy production by cancer cells even when oxygen is present (also referred to as aerobic glycolysis). It is hypothesized that this phenomenon leads to greater energy availability, more readily available biosynthetic intermediates, and resistance to apoptosis, all of which are beneficial for cancer cells to survive. Furthermore, the Warburg Effect is observed in many types of cancers, making it an attractive mechanism to target.5-7
Studies investigating the mechanistic underpinnings of the Warburg Effect have revealed novel, therapeutically relevant protein targets. Pyruvate dehydrogenase (PDH) is a pivotal enzyme located in the mitochondria that connects the process of glycolysis to the tricarboxylic acid (TCA) cycle. Further, PDK1 plays a pivotal role in metabolic reprogramming via phosphorylation of PDH and contributes to enhanced aerobic glycolysis (Warburg Effect).6,7 PDK1 is the main isoform of the PDKs; therefore, targeting PDK1 could be beneficial as an anti-glycolytic therapy for colorectal cancers. 8 Studies have shown that dichloroacetate (DCA) can reverse the Warburg effect by inhibiting the PDK protein that is present on the mitochondrial membrane. The inhibition of PDK1 results in the activation of pyruvate dehydrogenase complex (PDC); this allows for the influx of pyruvate into the mitochondria, which restores aerobic energy production. When DCA reactivates the mitochondria, apoptosis results from either the depolarization of the membrane or through upregulation of reactive oxygen species (ROS). 9 Despite the promising success of DCA in preclinical studies, clinical studies revealed some adverse events including neuropathy potentially due to the high dosage requirement.10-12 Additionally, DCA exhibited toxicity to neurons in rats, though the mechanism of this effect is not entirely clear. 13 Therefore, there is a need for the identification of safer compounds that can target PDK1 without causing such adverse effects. Studies have identified several other compounds capable of inhibiting PDK1. However, these drugs have faced challenges related to bioactivity, safety, and efficacy limiting their clinical applications.14,15 Therefore, we decided to identify a novel PDK1 inhibitor via a drug repurposing approach. Such approaches can help speed the drug development process if the identified compound has a proven safety profile. In our search for one such compound, we opted to employ a set of computational tools. Using a virtual ligand screening software, FINDSITEcomb, 16 we identified several compounds predicted to bind PDK1 with high affinity. We further pruned this list of compounds with the tool, DR. PRODIS, 17 to keep only those drugs with relatively few predicted off-target effects and side-effects. From this short list, we chose stearic acid (SA) as a candidate PDK1 inhibitor. SA is an 18-carbon saturated fatty acid found in various plant and animal fats, further bolstering our confidence in its safety. Previous experiments with the MDA-MB-231 breast cancer cell line revealed that SA induced apoptosis by activating caspases. 18 However, the current report is different in that it extends observations on inhibition of SA on 3 colorectal cell lines, provides new information on the mechanism of action of SA, and reports synergy in combination drug studies with 5-FU.
The present study was performed to investigate the inhibitory potential of SA on colorectal adenocarcinoma cells (DLD-1) as well as its mechanism of action. Further, combination studies with SA and 5-Fluorouracil (5-FU), which is a known chemotherapeutic that is currently used as treatment for colorectal cancer, were conducted to determine if synergy occurred. SA was also tested on other colon cancer cell lines, such as HCT 116 and Caco-2, and on the non-tumorigenic cell line-NIH-3T3.
Materials and Methods
PDK1 virtual ligand screening
First, we ran the virtual ligand screening software, FINDSITEcomb, 16 to identify compounds predicted to bind to PDK1. We selected resulting compounds with a score of over 0.9. Then we applied, DR. PRODIS, 17 to identify the subset of compounds from this list with a killing index of 0. SA was chosen from this final list, as it had a score of 0.911 and a killing index of 0.
Cell lines and compounds
Human colorectal adenocarcinoma (DLD-1), human colorectal carcinoma (HCT 116), human colorectal adenocarcinoma (Caco-2) and NIH-3T3 cell lines were obtained from the American Type Culture Collection (ATCC). Cell cultures were maintained at 37°C in an atmosphere of 5% CO2 and 95% air. Growth medium for DLD-1 cell line was prepared by supplementing RPMI with 10% FBS, 1% Penicillin/Streptomycin, and 2 mM L-Glutamine. HCT 116 cell line was maintained in McCoy’s 5A medium supplemented with 10% FBS, 1% Penicillin/Streptomycin, and 2 mM L-Glutamine. Caco-2 cell line was maintained in EMEM with 20% FBS and 1% Penicillin/Streptomycin antibiotic solution. SA and 5-FU were purchased from Sigma-Aldrich (S4751 and F6627, respectively) and were dissolved in DMSO to prepare the stock solutions. NIH-3T3 cells were grown in DMEM, supplemented with 10% FBS, 1% Penicillin/Streptomycin, and 2 mM L-Glutamine. CCK-8 was purchased from Bimake.
Assessment of cytotoxicity of SA on DLD-1, HCT 116, and Caco-2 cells
DLD-1 cells were plated in 96-well plates (5000 cells/well) in triplicate for the control and SA treatments. After a 24-hour incubation period, the medium was either replaced with fresh medium for the control cultures or replaced with medium containing varying concentrations of SA for the treatment groups. The concentrations tested were 100, 75, 50, 25, and 10 µM. To simulate hypoxic condition, 100 µM of cobalt chloride solution was added to the growth medium. After 72 hours of incubation, cell viability was assessed via a Cell Counting Kit-8 (CCK-8) assay. Cells (control and treated) from 72-hr experiment were re-seeded at a density of 500 cells/well in a 6-well plate. After allowing them to grow for 7 days in drug-free medium, colonies were fixed in methanol and stained with Crystal Violet.
Flow cytometry analysis
For deciphering the signaling pathway(s) impacted by SA, various flow cytometry assays were performed. Cells were plated in triplicate in 6-well culture plates (2 × 104 cells/well). After 24 hours of incubation, the media was replaced with fresh media for the control wells and with media containing 60 µM SA for the treatment group. After 72 hours, cells were harvested and processed as per the Muse cell kit protocols. All the assay kits were procured from Luinex. The flow cytometry assays performed include the Annexin V & Dead Cell assay (MCH100105), Multicaspase assay (MCH100109), Caspase-3/7 assay (MCH100108), MitoPotential assay (MCH100110), Bcl-2 Activation Dual Detection assay (MCH200105), and Cell Cycle Analysis (MCH100106). The cells were analyzed using a MUSE cell analyzer according to the manufacturer’s instructions (Luminex). Each assay was performed at least 3 times independently.
TMRE-mitochondrial membrane potential analysis
Cells were plated in four-chamber cell culture glass slides at a density of 20 000 cells/chamber. After a 24-hour incubation period, the medium was either replaced with fresh medium for the control cultures or replaced with medium containing 60 μM of SA for the treatment groups. After 72 hours of incubation, to detect the changes in mitochondrial membrane potential in live cells, cells were stained with TMRE-Mitochondrial Membrane Potential Assay Kit (Abcam—ab113852) as per manufacturer’s instructions. FCCP ((carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone) was used to treat control cells as a positive control.
Cytochrome C analysis
Cells were plated in 6-well plates at a density of 20 000 cells/well. After a 24-hour incubation period, the medium was either replaced with fresh medium for the control cultures or replaced with medium containing 60 μM of SA for the treatment groups. After 72 hours, cells were harvested and processed as per the Cytochrome C Human ELISA kit (Invitrogen KHO1051). Absorbance was measured using Infinite 200 pro (Tecan, USA) plate reader and data analyzed using GraphPad Prism software.
Inhibitory potential of SA in combination with 5-FU
To determine whether SA and 5-FU combination is synergistic and can be used to decrease the concentration of 5-FU currently used in clinical treatments; CCK-8 assays, flow cytometry assays, and clonogenic assays were performed for combinatorial studies with SA and 5-FU in DLD-1 cells. A dose response curve was first generated in DLD-1 cells for 5-FU using 100, 75, 50, 25, and 10 µM concentrations of the drug. The concentration of SA was then held constant at 60 µM, and the combination studies were conducted with 50, 25, 10, and 1 µM concentrations of 5-FU and the effect was assessed by CCK-8 assay. The flow cytometry and clonogenic assays for combination studies were done using 60 µM SA and 6 µM 5-FU. The procedures followed for the CCK-8, flow cytometry, and clonogenic assays were the same as described previously and each experiment was performed at least 3 times independently.
Assessment of SA toxicity in other cell lines
To determine the degree of toxicity of SA on other cancerous cell lines, a colorectal carcinoma cell line (HCT 116) and an epithelial colorectal adenocarcinoma (Caco-2) cell line and NIH-3T3 fibroblast cell line (non-tumorigenic) were utilized. CCK-8 assays were conducted on treatment with 60 µM SA.
Statistical analysis
The data were analyzed with an ordinary 2-way ANOVA using an alpha value of 0.05. Also, Sidak’s multiple comparisons 2-way ANOVA analysis was conducted with an alpha value of 0.05 The asterisks indicate significance at an alpha level of 0.05.
Results
SA inhibits the viability of DLD-1 cells
To evaluate the inhibitory potential of SA in the colorectal adenocarcinoma cell line—DLD-1, cells were treated with varying concentrations of SA for 72 hours under normoxic and hypoxic conditions. Treatment with SA resulted in dose-dependent inhibition with an IC50 of 60 µM under normoxic condition (Figure 1A). When same treatments were administered under hypoxic condition, as described in “Materials and Methods,” a dose-dependent inhibition was also observed with an IC50 of 60 µM (Figure 1B). Further, when DLD-1 cells were treated with 5-FU alone at various concentrations in the normoxic environment, a dose dependent inhibition was also observed with an IC50 of 25 µM as shown in Figure 1C and when assessed under the hypoxic condition, the IC50 was 30 µM (Figure 1D).

Cytotoxic effects of SA and 5-FU on colorectal adenocarcinoma (DLD-1) cells. Using CCK-8 assay, the cytotoxicity of SA on DLD-1 cells was determined in normoxic (A) and hypoxic (B) conditions. Means and standard deviations from 3 trials are shown. Cytotoxic effects of 5-FU on DLD-1 cells were determined using CCK-8 assay under normoxic (C) and hypoxic (D) conditions. The means and standard deviations from 3 trials are shown. The asterisks indicate significance at an alpha level of 0.05.
Combination of SA with 5-FU is synergistic
To assess the synergistic inhibitory potential of SA with 5-FU, DLD-1 cells were treated with varying concentrations of 5-FU with the addition of 60 µM SA for 72 hours under normoxic and hypoxic conditions. Combination of SA with 5-FU under normoxic condition resulted in an IC50 of 6 µM (Figure 2A) as compared to the IC50 of 25 µM when DLD-1 cells were treated 5-FU alone. Administration of combination treatments under hypoxic condition resulted in an IC50 of 1 µM (Figure 2B), as compared to the IC50 of 30 µM when 5-FU alone was used under hypoxic condition. Moreover, the inhibitory potential of the treatments was also tested via clonogenic assay (Figure 2C). Once again, the combination treatment of SA and 5-FU was most effective in deterring the ability of DLD-1 cell growth, when compared to the control. Further, control and treated cells were stained with phalloidin and DAPI and images were taken by confocal microscopy (Figure 2D). As can be seen in the images the treatment groups show enlarged as well as rounded cell morphology when compared to the control. The morphological changes observed are hallmarks of toxicity.

Combination studies conducted with SA and 5-FU in DLD-1 cells. Using CCK-8 assay, the effects of the combination of 60 µM SA and varying concentrations of 5-FU on DLD-1 cells were determined in normoxic (A) and hypoxic (B) conditions. Results from clonogenic assays (C) conducted with cells treated with SA, 5-FU, and their combination are depicted. Means and standard deviations from 3 trials are shown. The asterisks indicate significance at an alpha level of 0.05. (D) Images of cells stained with phalloidin, DAPI are depicted. Compared to control, the treatment groups show enlarged as well as rounded cell morphology, which are hallmarks of toxicity.
SA-induced inhibition of cell viability is mediated through programmed cell death pathway in DLD-1 cells
To understand the mechanism of action of SA inhibition of DLD-1 cell proliferation, flow cytometry assays were conducted. Cells were treated with 60 µM SA for the SA treatment group, 6 µM 5-FU for the 5-FU treatment group, and 60 µM SA with 6 µM 5-FU for the combination treatment group. Annexin V and Dead Cell flow cytometric assay were performed to determine the involvement of an extrinsic apoptosis pathway. When the profiles of the control (Figure 3A) and treatment groups: 60 µM SA, (Figure 3B) 6 µM 5-FU (Figure 3C), and combination of 60 µM SA with 6 µM 5-FU (Figure 3D) were compared, there was a significant increase in the percentage of cells in the apoptotic groups for the SA and combination treatment groups. While the 5-FU treatment group was similar to the control group, its combination with SA depicted increased apoptosis when compared to both SA and 5-FU alone (Figure 3E). The total apoptotic cells for each group of the study is shown (Figure 3F).

Annexin V Dead Cell flow cytometry assay to assess apoptosis. Scatter plots for the control cells (A), cells treated with 60 µM SA (B), cells treated with 6 µM 5-FU (C), and cells treated with the 60 µM SA plus 6 µM 5-FU combination (D) are shown. The average results from 3 independent estimations with standard deviations are depicted (E). The profile of 5-FU (C) was similar to that of the controls (A). Total apoptotic population in all the groups studied is represented in (F). The asterisks indicate significance at an alpha level of 0.05.
Caspases are involved in inducing apoptosis by SA
In order to decipher whether programmed cell death in DLD-1 treated cells involved multicaspase, multicaspase flow cytometric assay was performed. Compared to the control cell population (Figure 4A), in the combination studies, the caspase positive cell population was higher (Figure 4D). Quantitative estimations from multiple trials are summarized in Figure 4E. Total caspase positive cells in all the groups is represented in Figure 4D.

Assessment of multicaspase in DLD-1 cells on treatment with SA, 5-FU and SA+ 5-FU combination. Sample profiles for the control cells (A), cells treated with 60 µM SA (B), cells treated with 6 µM 5-FU (C), and cells treated with the 60 µM SA and 6 µM 5-FU combination (D) are shown. The average results from 3 assays with standard deviations are depicted (E). The data was analyzed with an ordinary 2-way ANOVA using an alpha value of 0.05, the combination treatment groups display increase in caspase positive cells when compared to the control. Total caspase positive cells in each group is represented in (F). The asterisks indicate significance at the alpha level of 0.05.
Flow cytometry of caspases 3/7 in DLD-1 cells
To further elucidate the mechanism of action of SA, Caspase 3/7 flow cytometry assay was performed to analyze the apoptotic status of the DLD-1 cells based on activation of the executioner caspase 3/7. Compared to the control population, (Figure 5A), there was no statistically significant increase in caspase 3/7. Quantitative estimations from multiple trials are summarized in Figure 5E. Total caspase 3/7 positive cells in all the groups analyzed are represented in Figure 5F.

Caspase 3/7 flow cytometry assay to determine the impact of SA, 5-FU and combination treatments on DLD-1 cells. Representative scatter plots for control cells (A), cells treated with 60 µM SA (B), cells treated with 6 µM 5-FU (C), and cells treated with the 60 µM SA and 6 µM 5-FU combination (D) are shown. The average results from 3 assays with standard deviations are also depicted (E). Total caspase 3/7 positive cells in all the groups are represented in (F). The data was analyzed with an ordinary 2-way ANOVA using an alpha value of 0.05. There was no statistically significant difference when compared to the control.
A modest increase in total depolarized mitochondria was observed on treatment with SA and in combination with 5-FU
To investigate further the mode of action of SA, 5-FU and combination of SA with 5-FU; MitoPotential flow cytometric assays were performed. Representative scatter plots of the control sample Figure 6A, SA (60 µM) treated sample (B), 5-FU (6 µM) treated group (C), and combination group (SA-60 µM plus 6 µM of 5-FU) are shown (D). Average percent gated profile of the MitoPotential in the samples analyzed is shown in (E). Total depolarized cells in all the groups analyzed are represented in (F). A modest increase in the total depolarized mitochondrial membrane potential was observed in the SA treated group. This change although modest was maintained on combination with 5-FU.

MitoPotential flow cytometry assay depicts that SA induces a modest depolarization of the mitochondrial membrane. Assays for the control cells (A), cells treated with 60 µM SA (B), cells treated with 6 µM 5-FU (C), and cells treated with the 60 µM SA and 6 µM 5-FU combination (D). Percent gated populations in the samples analyzed are represented in (E). An average of total depolarized cells in all the groups analyzed is represented in (F).
SA treatment affects Bcl-2 in DLD-1 cells
Bcl-2 is a key player in the survival and proliferation of various cancers. To investigate the effect of SA, 5-FU, and their combination on Bcl-2 levels, a Bcl-2 flow cytometric assay was conducted. The SA treatment group showed increased non-expression of Bcl-2 molecules and decrease in the activated Bcl-2 when compared to the control group. The 5-FU treatment group also depicted increased non-expression and decrease in activated Bcl-2 as compared to the control, but the SA treatment group showed a higher percentage of non-expressing cells. The combination treatment group was similar to that seen with the SA group. Therefore, the effect observed with SA alone on Bcl-2 was maintained in the combination studies (Figure 7).

Bcl-2 levels and expression profile depict that SA induces inactivation of Bcl-2 and the SA plus 5-FU combination is synergistic. SA induces Bcl-2 inactivation, decrease in activated molecules and a significant increase in the percentage of non-expressing cells. Assays for the control cells (A), cells treated with 60 µM SA (B), cells treated with 6 µM 5-FU (C), and cells treated with the 60 µM SA and 6 µM 5-FU combination (D) are shown. The average results from 3 assays with standard deviations are depicted (E). The data was analyzed using an ordinary 2-way ANOVA with an alpha value of 0.05. The asterisks indicate significance at the alpha level.
SA exhibited maximum inhibition of DLD-1 cells in comparison to other colon cancer cells
Data from further studies conducted on HCT 116 and Caco-2 cells, revealed that SA had maximal inhibitory effect on DLD-1 cells. The inhibitory activity of SA for the colon cancer cell lines tested was DLD-1 > Caco-2 > HCT 116. Furthermore, at the concentration tested, SA had minimal effect on NIH-3T3—a non-cancerous cell line (Figure 8).
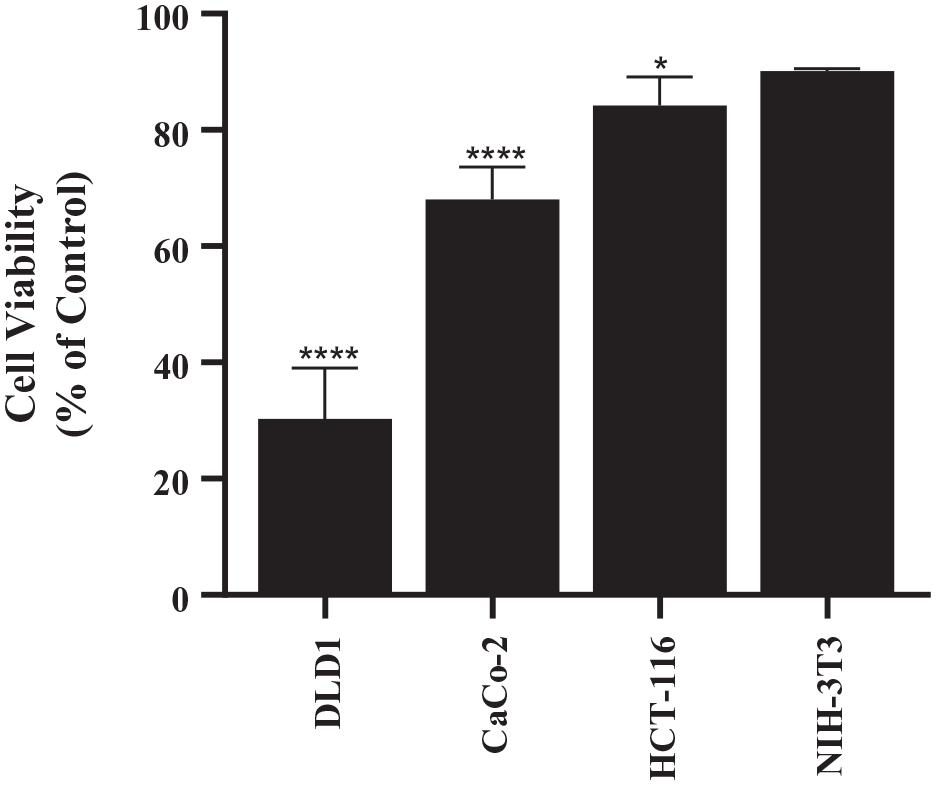
Differential sensitivity of colon cancer cell lines and non-tumorigenic cell line to SA. The cell lines were cultured in their respective media, as described in Section 2.2, and cells were plated in 96-well plates (5000 cells/well). After 24 hours, the cells were treated with 60 μM SA in their respective medium. After 72 hours of treatment, CCK-8 assay was conducted to determine cell viability. Mean ± standard deviation of triplicates is represented. Sidak’s multiple comparisons 2-way ANOVA analysis was conducted with an alpha value of 0.05, each cell line was compared to its corresponding control. DLD-1 and Caco-2 cells exhibited significantly more toxicity to SA than did HCT 116 or NIH-3T3.
Discussion
There are drugs currently being used to manage colorectal cancer in the clinic; however, due to their limited efficacy and drug resistance, there is an imminent need for more effective, novel treatments. We opted to target the glycolytic pathway in cancer cells because these cells have reprogrammed their metabolic pathway for achieving the energy required for their uncontrolled growth. On these lines, a potential strategy being investigated is the inhibition of PDK1—a pivotal enzyme connecting glycolysis to the Krebs’s cycle. 8 Therefore, inhibiting PDK1 would curb the energy source of colorectal cancer cells and induce cell death. To find a PDK1 binding molecule, we opted to use the FINDSITEcomb virtual ligand screening software. 16 Using this software, SA was identified as a potential PDK1 inhibitor. SA was also predicted to have minimal off-target effects, making it a potential chemotherapeutic candidate for colorectal cancers. 17 SA has only been previously tested on the MDA-MB-231 breast cancer cell line, and SA was reported to induce apoptosis by activating caspases. 18 Our experimentation with SA on colorectal adenocarcinoma cells (DLD-1) also demonstrated involvement of caspases as observed in MDA-MB-231 breast cancer cell line. Currently, 5-FU is the drug of choice for colorectal cancers, however efficacy and resistance is a factor to be addressed.19-22 To this end, we opted to perform synergy experiments with SA and 5-FU. Synergy was revealed when SA was combined with 5-FU as shown by the notable decrease in IC50 of 5-FU from 25 to 6 µM in combination when compared to 5-FU alone. Further, clonogenic assay showed that short term treatment with SA impacted the long-term proliferation. When SA was tested on other cancerous cell lines, including Caco-2 and HCT 116, it was found that SA exhibited differential sensitivity. The epithelial colorectal adenocarcinoma cell line, DLD-1, was the most sensitive to SA while another colorectal adenocarcinoma cell line, Caco-2, exhibited less sensitivity. HCT 116, which is an epithelial colorectal carcinoma cell line, exhibited the least sensitivity to SA. Studies with NIH-3T3, a non-cancerous fibroblast cell line, confirmed that SA had minimal inhibitory effect on normal cells suggesting potentially fewer side effects. The difference in tissue type could have also contributed to the differential response of NIH-3T3 to SA: fibroblast (NIH-3T3) vs epithelial (colon cancers). With regard to the colon cancer cells; we envision that this could be due to the p53 status in HCT 116 and DLD-1. HCT 116 has wild type p53, whereas DLD-1 has mutant p53. Various factors could contribute to the Warburg effect; notably, it is reported that tumor-associated mutant p53 can control this effect. 23
Interestingly, mechanism of action of SA elucidated through flow cytometry assays revealed that the inhibitory action was through the extrinsic apoptotic pathway involving multicaspases. Further, SA also impacted Bcl-2 which plays a pivotal role in the control of cancer cell survival and proliferation. The levels of this entity were affected with an increase in the inactivated molecules and decrease in activated levels accompanied by increase in the non-expressing cells in DLD-1. Importantly, this effect was maintained in the combination studies with 5-FU. Similarly, the significant effect of SA on the extrinsic programmed cell death pathway which led to an increase in caspases was also maintained in the combination experiments with 5-FU. Thus, we envision that the combination of SA with 5-FU would be an efficacious treatment for combating colorectal adenocarcinomas.
The downregulation of Bcl-2 and observed apoptosis with SA treatment are consistent with the hypothesis of PDK1 inhibition by SA, although more mitochondrial depolarization would have been expected. The retention of cell viability in the non-tumorigenic cell line when treated with SA is also consistent with PDK1 inhibition, as we would expect minimal toxicity to normal cells. Despite these findings, more work is needed to definitively determine if the mechanism of action of SA is through binding to PDK1 or other mediators.
Future studies should be conducted to explore the effects of SA in combination with 5-FU in vivo to further determine its efficacy and safety. Additionally, inhibitory efficacy of SA alone and in combination with 5-FU needs to be determined against 5-FU resistant colon cancer cells. Further, hypoxic regions exist in the tumor microenvironment due to increased cell proliferation. 24 This hypoxic environment causes the cancer cells to be less responsive to radiation and chemotherapy. 25 Therefore, compounds that can cause cell death in hypoxic conditions are essential. There are several studies that indicate the benefits of inhibiting PDK1 including under hypoxic tumor environments. 26 Our findings of the significant inhibitory effect of SA alone and in combination with 5-FU in DLD-1 cells under hypoxic environment as well could be additionally beneficial for efficaciously eradicating colorectal cancer cells. Furthermore, combination of immunotherapy entities such as Cetuximab with SA and 5-FU could also be considered because it has been reported that Cetuximab, an EGFR blocking antibody in addition to its target-induced effects, sensitizes the cells to ROS production in head and neck squamous carcinoma cells (HNSCC). Notably, PDK1 knockdown or inhibiting PDK1 by dichloroacetic acid (DCA) and then combining with Cetuximab led to enhanced ROS production and marked reduction in tumor growth in Cetuximab resistant HNSCC. 27 Furthermore, discovery of inhibitors targeted against PDK-1 has been an active area of research.28,29 Thus, future in vivo studies with SA and 5-FU are warranted, and this could lead to novel treatment options for colorectal carcinomas.
Conclusion
We envision that the significant decrease in the IC50 of 5-FU could translate into minimal side effects of 5-FU and increase the efficacy of the treatment due to the multifaceted action of SA. The data generated from the current studies on the inhibition of colorectal adenocarcinoma by SA discovered by the use of the computational program as well as synergistic action with 5-FU should open up novel therapeutic options for the management of colorectal carcinomas.
Supplemental Material
sj-docx-1-cix-10.1177_11769351211065979 – Supplemental material for Computational Identification of Stearic Acid as a Potential PDK1 Inhibitor and In Vitro Validation of Stearic Acid as Colon Cancer Therapeutic in Combination with 5-Fluorouracil
Supplemental material, sj-docx-1-cix-10.1177_11769351211065979 for Computational Identification of Stearic Acid as a Potential PDK1 Inhibitor and In Vitro Validation of Stearic Acid as Colon Cancer Therapeutic in Combination with 5-Fluorouracil by Jonathan Mitchel, Pratima Bajaj, Ketki Patil, Austin Gunnarson, Emilie Pourchet, Yoo Na Kim, Jeffrey Skolnick and S. Balakrishna Pai in Cancer Informatics
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Wallace H. Coulter Department Biomedical Engineering at Georgia Institute of Technology and Emory University and NIH grant GM-118039 to Dr. Skolnick. Yoo Na Kim received funding for research from Kern Family Foundation grant. We would also like to acknowledge the support from Coulter Fund and Georgia Tech Technology Fee.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Jonathan Mitchel, Pratima Bajaj and Ketki Patil contributed equally to this work.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.