
Abstract
Introduction:
The emergence of new omics approaches, such as genomic algorithms to identify tumor mutations and molecular modeling tools to predict the three-dimensional structure of proteins, has facilitated the understanding of the dynamic mechanisms involved in the pathogenesis of low-grade gliomas including oligodendrogliomas and astrocytomas.
Methods:
In this study, we targeted known mutations involved in low-grade gliomas, starting with the sequencing of genomic regions encompassing exon 4 of isocitrate dehydrogenase 1 (IDH1) and isocitrate dehydrogenase 2 (IDH2) and the four exons (5-6 and 7-8) of TP53 from 32 samples, followed by computational analysis to study the impact of these mutations on the structure and function of 3 proteins IDH1, IDH2, and p53.
Results:
We obtain a mutation that has an effect on the catalytic site of the protein IDH1 as R132H and on the catalytic site of the protein IDH2 as R172M. Other mutations at p53 have been identified as K305N, which is a pathogenic mutation; R175 H, which is a benign mutation; and R158G, which disrupts the structural conformation of the tumor suppressor protein.
Conclusion:
In low-grade gliomas, mutations in IDH1, IDH2, and TP53 may be the key to tumor progression because they have an effect on the function of the protein such as mutations R132H in IDH1 and R172M in IDH2, which change the function of the enzyme alpha-ketoglutarate, or R158G in TP53, which affects the structure of the generated protein, thus their importance in understanding gliomagenesis and for more accurate diagnosis complementary to the anatomical pathology tests.
Introduction
Low-grade gliomas are a group of heterogeneous brain tumors with distinct biological and clinical properties; in the past, subgroups and classification of gliomas were defined by histological features. However, this classification method does not reflect tumor heterogeneity, does not allow a precise diagnosis, and is not a reliable method for treatment.
In the past 3 years, new discoveries of molecular biomarkers and diagnosis in adults with low-grade gliomas have resulted in a change in pathological classification of all gliomas such as astrocytomas and oligodendrogliomas.1,2 World Health Organization’s updated classification of central nervous system tumors in 2016 insists that the standard diagnostic evaluation of low-grade gliomas should now include a molecular assessment of the genes involved, like the mutations of isocitrate dehydrogenases (IDHs) IDH1 and IDH2 and p53 protein.3,4
The IDH catalyzes the conversion of isocitrate to alpha-ketoglutarate (KG). The IDH is present in 3 isoforms: IDH1 located in the cytoplasm, IDH2 located in the mitochondria, and IDH3 which is part of the tricarboxylic acid cycle. 4
Patients with low-grade gliomas including astrocytomas and oligodendrogliomas are characterized by mutations in the active site of IDH1 at position R132 and IDH2 at position R172. In the mutated case, both IDH1 and IDH2 catalyze the conversion of alpha-KG to beta-hydroxyglutarate (2-HG); supernormal levels of intracellular 2-HG result in hypermethylation of target genes such as TET2 involved in cell differentiation. 4 The wild-type TP53 gene has an indispensable role in several cellular processes, including tumor suppression, apoptosis, DNA repair, autophagy, and so on, but is often mutated or deleted at the early stage of low-grade gliomas. In about 30% to 40% of cases of astrocytomas, a loss of p53 function can be observed at an early stage. Mutations of p53 in certain residues such as R158G can disrupt the structural and functional properties of the protein to varying degrees and affect the prognosis of patients. 5
This study aims to identify mutations in the biomarkers that are the most implicated in low-grade gliomas—IDH1, IDH2, TP53—and study the effect of these mutations on the function and structure of the proteins.
Materials and Methods
Ethics Statement
This study was approved by the Ethics Committee of Biomedical Research of Mohammed V University on the grounds of satisfactory conditions of validity, scientific relevance, research interests, ethical relevance, satisfactory conditions of human pretensions, intelligibility of the note of information, and conformity of the modalities of collection of consent. The Ethics Committee’s deliberations are based on the Helsinki Declaration (2008 version), the International Ethical Guidelines for Biomedical Research Involving Human Subjects of the Council for International Organizations of Medical Sciences (CIOMS, 2002 version), and the National Law (no. 28-13; 2015) for the Protection of Persons Participating in Biomedical Research.
Written informed consent was obtained from all participants prior to blood collection, and all of them were informed that blood samples would be used for research projects.
Study design and collection of samples
This pilot study was carried out on 32 samples, including 24 tumors and 8 controls.
For the 24 tumors, we have 20 paraffin-embedded tissue blocks (14 astrocytomas and 6 oligodendrogliomas) and 4 blood samples (2 astrocytomas and 2 oligodendrogliomas).
For the 8 control samples, we have 6 paraffin-coated tissue blocks belonging to the same patients who have tumor, which was cut from the healthy part of each patient (3 astrocytomas and 3 oligodendrogliomas), and 2 blood samples independent of the other patients.
Twenty paraffin-embedded tissue blocks of patients between 2013 and 2017 were collected retrospectively from the Department of Pathological Anatomy, Specialty Hospital, CHU Ibn Sina, Rabat.
Five milliliters of peripheral blood was collected in tubes containing ethylenediaminetetraacetic acid from 4 patients, after informed consent was provided by patients through institutional review board–approved protocols, from the Department of Neurosurgery, Specialty Hospital, CHU Ibn Sina, Rabat, and they were treated in a prospective manner.
DNA preparation, polymerase chain reaction amplification, and sequencing
Twenty-six (20 patients, 6 controls) formalin-fixed paraffin-embedded (FFPE) tissue blocks were prepared by a semi-automatic microtome (HM 340E; Thermo Fisher Scientific), 8 µM sections were cut sequentially from each block, and each section was collected in a separate slide. For each slide, 25 mg of tissue was scraped and then placed in a 1.5-mL tube to extract DNA using the phenol-chloroform method.
Dewaxing was performed with xylene (1 mL); after that, several ethanol (1 mL) washes were carried out.
Cell lysis was performed by the addition of 300 µL of extraction buffer. The digestion and the elimination of proteins were carried out, respectively, by adding 30 µL of Proteinase K (10 mg/mL) and 300 µL of phenol-chloroform; 250 µL of ammonium acetate was used to precipitate the DNA. Concerning the 6 blood samples, MyTaq Blood-PCR Kit (BIO-25054) was used directly to perform polymerase chain reaction (PCR) without the DNA extraction step.
DNA quantification was assessed with QuantiFluor dsDNA System (Promega E4871); samples were also evaluated for quality using electrophoresis.
To identify IDH1, IDH2, and TP53 mutations, the genomic regions encompassing exon 4 of IDH1 and IDH2 and the four exons (5-6 and 7-8) of TP53 were designed by Primer3 (PCR primer design tool version 2.0). The following sequences of primers were used: IDH1 forward, 5′-TGATGAGAAGAGGGTTGAGGA-3′ and reverse, 3′-ATCCCCATAAGCATGACGAC-5′; IDH2 forward, 5′-CAAGCTGAAGAAGATGTGGAA-3′ and reverse, 3′-CAGAGACAAGAGGATGGCTA-5′; fifth and sixth exons of TP53 forward, 5′-AATGGTTCACTGAAGACCCA-3′ and reverse, 3′-AAAAGTTGACACGTTATTCAATT-5′; and seventh and eighth exons of TP53 forward, 5′-TAGGTTGGCTCTGACTGTA-3′ and reverse, 3′- AAGTTGAATGTTATAAAAGTT-5′.
About 5 to 50 ng of DNA was used to perform the PCR (SimpliAmp Thermal Cycler A24811) with 1 µL of each primer and 12.5 µL of MyTaq Mix (Bioline, BIO-25041). The PCR conditions for all sequences are as follows: 30 cycles with denaturing at 95°C for 30 seconds; annealing for 40 seconds at 51°C, 50°C, 52°C, and 54°C for IDH1, IDH2, fifth and sixth exons of TP53, and seventh and eighth exons of TP53, respectively; and the final extension at 72°C for 40 seconds.
The PCR products were purified using the ExoSAP-IT Express PCR Product (75001.1.ML; Thermo Fisher Scientific). The sequencing reaction was subjected to 25 cycles of amplification consisting of denaturation at 96°C for 10 seconds, annealing at 50°C for 5 seconds, and extension at 60°C for 4 minutes in a total volume of 10 µL. The purified product is sequenced using the BigDye X-terminator v3.1 Cycle Sequencing Kit (catalog number 4376486; Applied Biosystems) according to the supplier’s protocol.
Computational analysis
To understand the effects of mutations found on the structure and function of the protein, we used PolyPhen-2 Version 2.0.23 to predict the functional significance of substitution6,7 and Iterative Threading Assembly Refinement (I-TASSER) server version 5.1 for automated protein structure prediction to model protein structures for specific translated mutant exon sequences for all the mutations: R132H in IDH1, R172M in IDH2, and R158G, R175H, and K305N in TP53.8-10 I-TASSER use multiple alignments to match the first model to all structures in the Protein Data Bank (PDB) library.11,12 To select the final models, I-TASSER uses the SPICKER program to cluster all the structures based on similarity. 13 The confidence of each model is quantitatively measured by C-score that is calculated based on the significance of threading template alignments and the convergence parameters of the structure assembly simulations. Template modeling score (TM-score), root-mean-square deviation of atomic positions, and normalized B-factor are estimated based on C-score and protein length following the correlation observed between these qualities.14,15 Structural validation of the protein model was done by PROCHECK program 3.5.4 which determines the stereochemical aspects along with the main chain and side chain parameters with comprehensive analysis. The model was validated using the Ramachandran plot. It offers a simple view of the conformation of a protein and also displays a visualization of the energetic regions, for which we find the percentage of each region: the favored region, the allowed region, and the outlier region. These regions must be within a specified interval for validation of the protein structure.16,17
Protein packing was described using weighted contact number (WCN), and the sequence-specific conservation scores were computed following the protocol of ConSurf. The reciprocal of the WCN (rWCN) profile is used to compare with the sequence conservation profile,18,19 and then the visualization of the three-dimensional (3D) structure was performed using PyMOL Version 2.3.1. 20 SnapGene viewer software version 4.1.9 was used to assist with the interpretation of the sequence electropherograms generated by sequencing. 21
Results
All the samples included in the study were anonymized, and no information on the identity of any individual was available during analysis.
An R132H mutation of the IDH1 gene was detected in 11 out of 32 samples, including 9 astrocytomas and 2 oligodendrogliomas (Table 1). The R172M mutation in IDH2 was detected in 1 oligodendroglioma, and the R158G, R175H, and K305N mutations of the TP53 gene were identified in 5 astrocytomas. Table 1 shows the impact of amino acid substitutions on the structure and function of 3 proteins predicted by PolyPhen-2 (Figure 1). The PolyPhen-2 score represents the probability that a substitution is damaging, with score ranging from 0.0 (tolerated) to 1.0 (deleterious). PolyPhen-2 uses 2 databases to form the complete model. The first one is HumDiv, which is used to evaluate the rare alleles of loci potentially involved in complex phenotypes and includes all harmful mutations with known effects on diseases in UniProtKB databases. The second is HumVar, which contains all common human single nucleotide polymorphisms and nondamaging proteins. 22 All the IDH1 R132H, IDH2 R172M, and TP53 K305N, R158G, and R175H mutations present a score more than 0.5, so these variants are predicted to be damaging; however, R175H in p53 is a benign mutation.
Mutations of IDH1, IDH2 and TP53 genes.
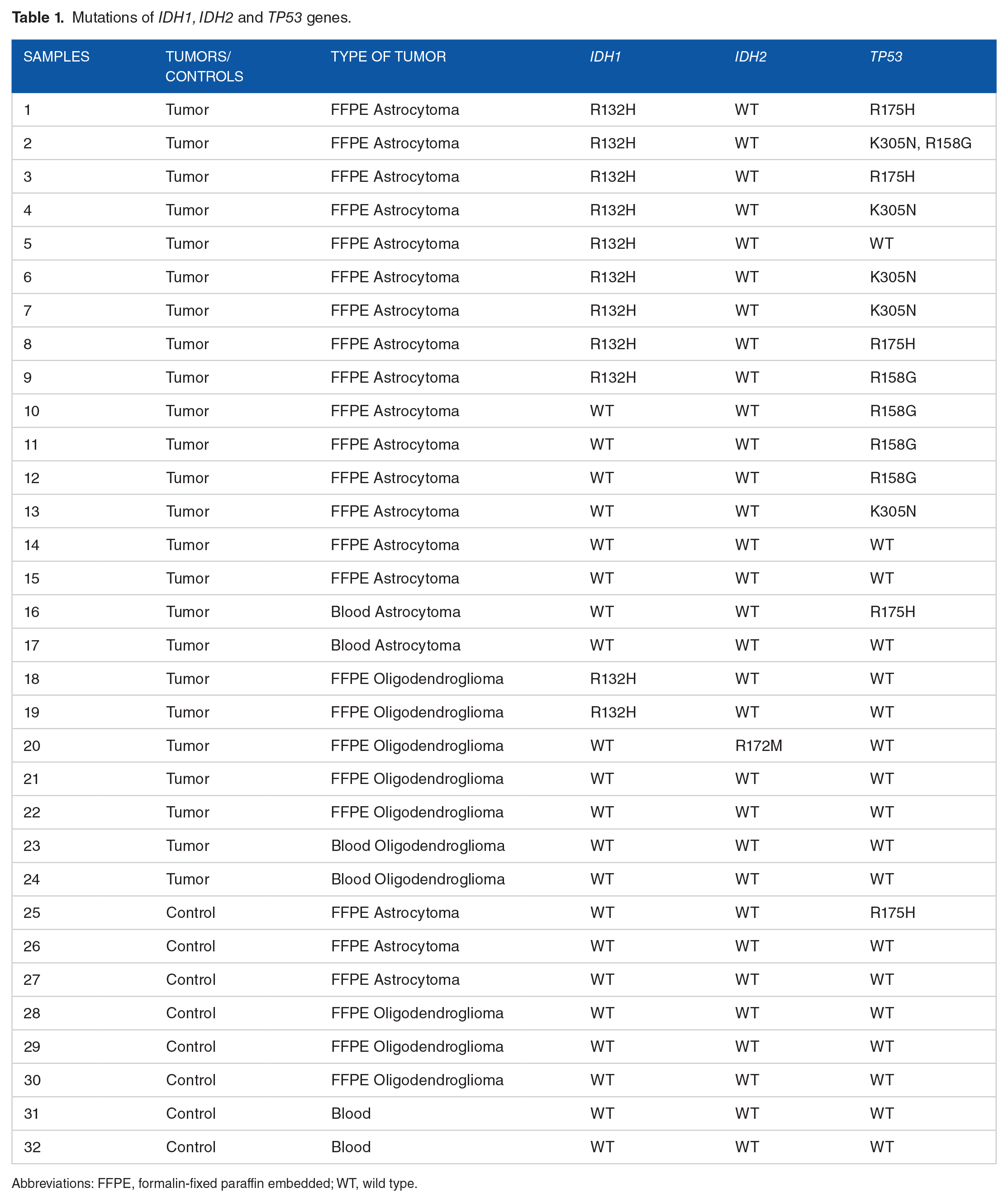
Abbreviations: FFPE, formalin-fixed paraffin embedded; WT, wild type.

PolyPhen-2 prediction of the possible effects of IDH1, IDH2, and TP53 mutations.
Protein Structure Prediction Server (PS) 2 Version 3.0 was used for calculating conservation scores. The series of conservation scores of a sequence is called conservation profile. In particular, in the conserving profile of a protein, the residue of a lower conservation score is more conserved than that of a higher conservation score. 23
Indeed, Figure 2 shows that the rWCN profile and the conservation profile overlap extremely well in the IDH1, IDH2, and p53 proteins. Here, we show that among the pathogenic mutations, these residues (IDH1 R132H, IDH2 R172M, and p53 R175H) are conserved and located in more compact environments, so it is reasonable to assume that these mutations are strongly associated with the functionality and the structure of the protein.

The rWCN profile (blue line) and ConSurf profile (black line) of the IDH protein. The mutated (A) IDH1, (B) IDH2, and (C) TP53 genes are marked in red circle. Both the rWCN and the conservation scores are normalized to their respective scores. Normalized B-factor profiles from I-TASSER for (D) IDH1, (E) IDH2, and (F) TP53.
In contrast, p53 K305N (Figure 2C) is located in a less conserved area because it has a positive score. The other part (Figure 2D to F) presents a value to indicate the extent of the inherent thermal mobility of the residues/atoms in the proteins, as well as the local precision of the residues in the strand, the helix, or the coil. The IDH1 R132H and p53 R158G mutations are found in the strand portion, and the IDH2 R172M, R175H, and K305N mutations are found in the coil position.
To estimate the quality of the models predicted by I-TASSER, C-score is calculated according to the significance of threading template alignments and the convergence parameters of the structure. It is typically in the range of [−5, 2]: the C-score of IDH1 is 1.31, of IDH2 is −0.84, and of p53 is −2.58.
For TM-score, which is a recently proposed scale for measuring the structural similarity between structures, this cutoff does not depend on the protein length. A TM-score >0.5 indicates a model of correct topology like IDH1 TM = 0.90 ± 0.06, IDH2 TM = 0.61 ± 0.14, and p53 TM = 0.42 ± 0.14; however, in the case where the TM-score is <0.17, there is random similarity.23,24
The modeled proteins are further validated by the Ramachandran plot generated by Procheck. The Ramachandran plot value for IDH1 (Figure 3A) was found to be 91.8% with 628 residues lying in the favored region, 7.7% of the residues lying in the additional allowed region, and 0.4% lying in the generously allowed region. No residues are located in the disallowed region. The number of glycine residues is 58 and the number of proline residues is 24. 25
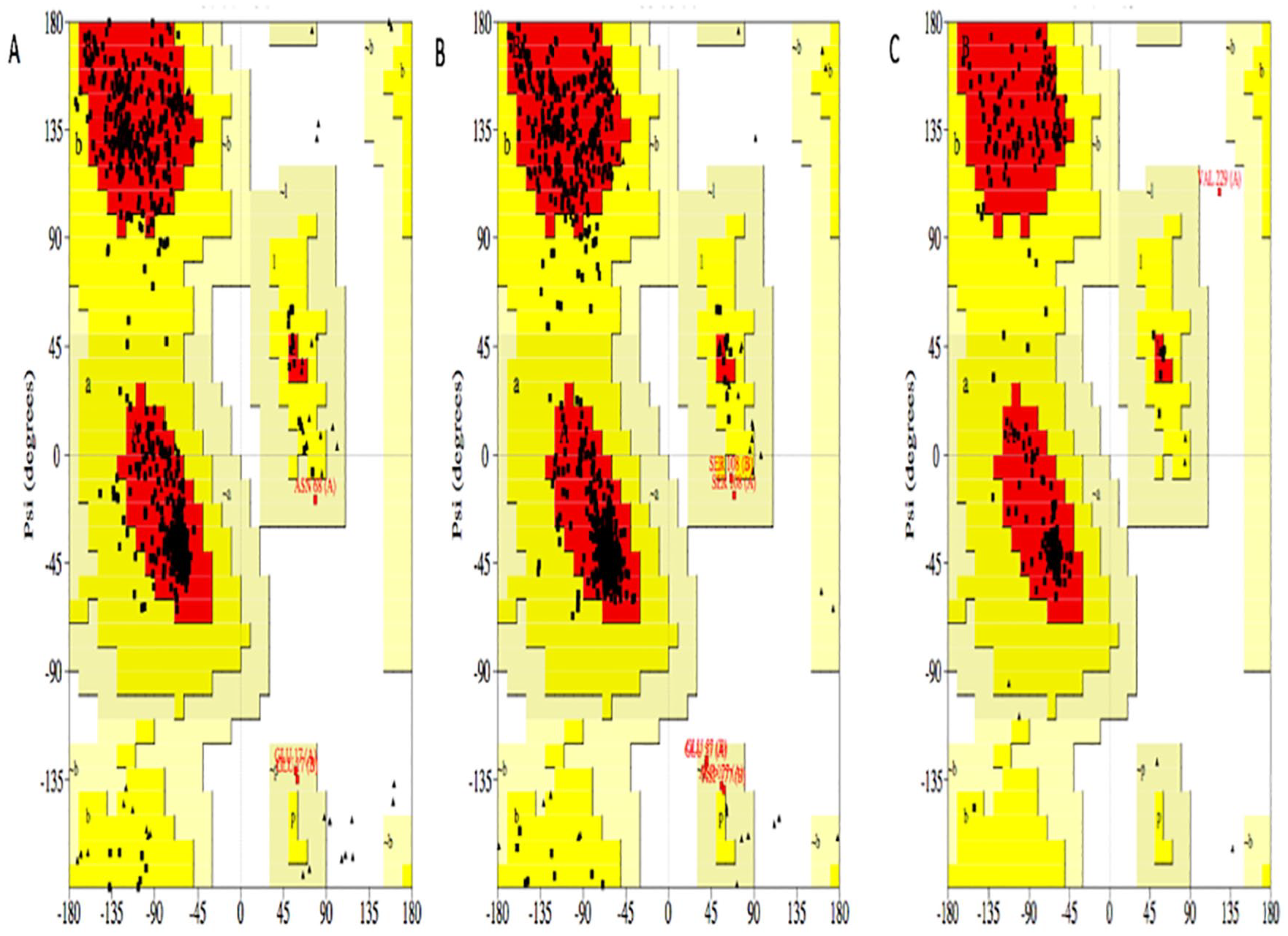
Ramachandran plot of (A) IDH1, (B) IDH2, and (C) p53 model. The core region, allowed region, and general region are colored with red, yellow, and beige, respectively.
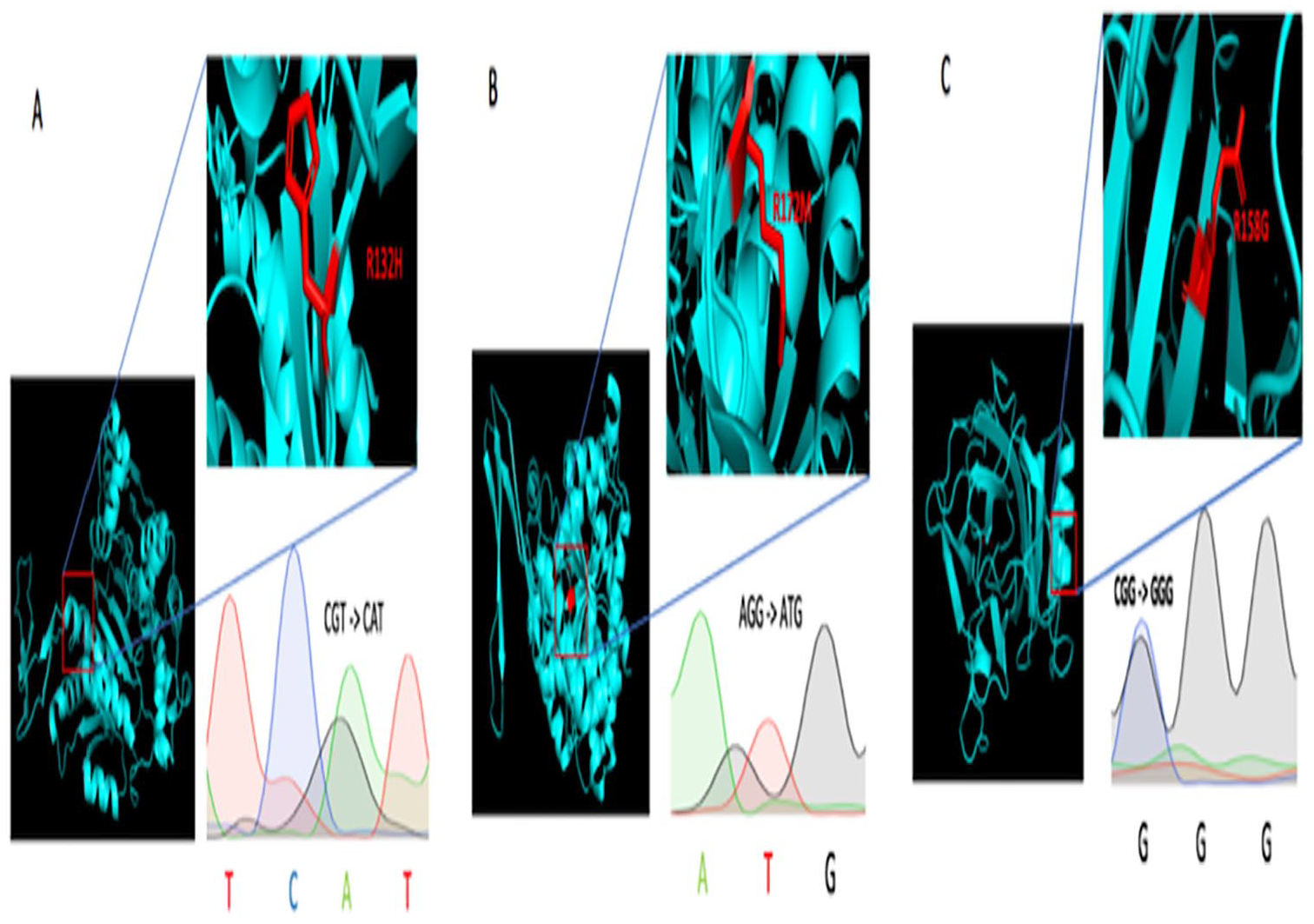
Chromatograms of mutated IDH1, IDH2, and TP53 gene sequences and structural analysis of proteins bearing the (A) R132H, (B) R172M (B), and (C) R158G mutations.
Figure 3B shows the plot value for IDH2 to be 91.9% with 647 residues lying in the favored region, 7.2% of the residues lying in the additional allowed region, and 0.9% lying in the generously allowed region. No residues are located in the disallowed region. The number of glycine residues is 66 and the number of proline residues is 30.
For p53 (Figure 3C), the plot value was found to be 93.1% with 202 residues lying in the favored region, 6.5% of the residues lying in the additional allowed region, and 0% lying in the generously allowed region. Only about 0.5% of the residues were located in the disallowed region. The number of glycine residues is 15 and the number of proline residues is 9. 25 Then the models generated by I-TASSER in PDB format were visualized using PyMOL. 26
Discussion
Recently, new biomarkers involved in low-grade gliomas have led to a change in the pathological classification.
Moreover, several studies were highlighted by the computational approach of the impact of mutations in IDH1 like I147S, V444A, and D375Y and in IDH2 like N439D and R140G27-29; using the same mechanism by which we carried out the sequencing of IDH1, IDH2 and TP53 genes, we were able to identify a few different amino acid substitutions in the IDH1, IDH2, and p53 proteins, such as IDH1 R132, IDH2 R173M, and p53 R175H, R158G, and K305N.6,30
The IDH mutations (Figure 3A and B) occurring in the catalytic site are likely to cause a loss of catalytic activity; these mutated genes encode for D2HG which is proposed to be an oncometabolite in the cytoplasm for IDH1 and in the mitochondria for IDH2; it competitively inhibits KG-dependent enzymes, including the TET family of 5-methylcytosine hydroxylases and the JmjC family of histones lysine demethylases, resulting in cell differentiation.27,28,31,32 Thirumal Kumar et al27,28 reveal that the drug therapeutics for D-2-hydroxyglutarate dehydrogenase are very limited; therefore, understanding the nature of molecular structure caused by these mutations will serve as platform for the development of novel targets for new drug therapy for D-2-hydroxyglutaric aciduria.
Concerning the mutation of p53 R158G (Figure 3C), arginine substitution on glycine at position 158 of p53 affects the structural conformation of the protein and may prevent DNA binding. This residue change is very frequent in human cancers such as gliomas.5,33
Anasuya Pal et al 34 investigated the involvement of TP53 mutations in breast cancer and found that most of the TP53 point mutations occur in the DNA binding domain and can be classified as DNA contact or structural mutations, such as R248W, and 2 structural mutants Y234C and H179R are resistant to apoptosis.
In our study, most of these mutations (IDH1 R132, IDH2 R173M, and p53 R175H, R158G, and K305N) have been identified in grade II and III glioma samples, suggesting a possible synergistic role in gliomagenesis.
However, Borger et al 29 have identified for the first time a high frequency of mutations in the IDH1 and IDH2 genes in cholangiocarcinomas specifically of intrahepatic origin, which indicates that IDH1 and IDH2 are involved in other types of cancer. Computational analysis showed that the mutations of IDH1 and IDH2 were probably harmful as well as the 2 mutations of the p53 R158G and K305N; on the contrary, the substitution R175H is a benign mutation (Figure 1). These results were calculated by the use of PolyPhen-2, which is a tool for predicting the possible impact of amino acid substitution on the function of a human protein (Figure 1). On the contrary, Thirumal Kumar et al 27 used SIFT as tools for predicting the pathogenicity of mutation found.
The prediction is based on a number of features comprising the sequence, and the phylogenetic and structural information characterizing the substitution. For a given amino acid substitution in a protein, we have extracted various sequence and structure based features of the substitution site and feed them into a probabilistic classifier. 35
Early effects of poor prognosis of p53 hotspot mutations have been demonstrated in low-grade astrocytomas and oligodendrogliomas. 36 The structural analysis of these p53 R158G mutants revealed their possible influence on tumor progression by disrupting the structure of the protein, especially as this mutation occurs in a very dense zone in the protein, as it was demonstrated using ConSurf, also used by Thirumal Kumar et al to calculate the protein conservation score. This type of mutation that stabilizes the DNA binding structure is called “structural mutants”; it affects the overall architecture of the DNA binding surface and modifies the conformation of the protein, unlike the wild-type p53 protein that acts as a tumor suppressor due to its DNA binding activity. 18 Finally, although we demonstrate detection of known IDH1, IDH2, and p53 mutations, limitations of this study include the difficult manipulation due to the shelf life of the samples included in the FFPE. Furthermore, the spectrum of biomarkers studied must be broadened in future studies to include the TERT promoter mutations and 1p/19q co-deletion to provide further value to the study and its attempt to understand gliomagenesis. 37
The coupling of new molecular methods with anatomical pathology analysis is thought to be part of daily clinical care as an essential tool for the diagnosis of low-grade gliomas and personalized systems medicine. It provides more accurate information and can also reduce the need for additional surgeries and provide important information for further radiotherapy or chemotherapy treatment as long as the treatment varies depending on the diagnosis. Despite these advances, it is important to note that much remains to be done in this area, particularly to develop innovative and improved computer models for so-called translational research.38,39
Conclusion
The discoveries of the past decade have completely changed our view of the genomics of human gliomas. Through computational analysis, we were able to study the pathogenicity of the IDH1, IDH2, and TP53 mutations and model the proteins generated in 3D, which allowed us to better understand gliomagenesis. These computer techniques will complement the analysis of anatomical pathology, which will improve diagnosis of astrocytomas and oligodendrogliomas.
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was carried out under National Funding from the Moroccan Ministry of Higher education & Scientific Research (PPR program) to A.I. This work was also supported by a grant from the National Institutes of Health for H3Africa BioNet to A.I. and Research Institute of the Foundation Lalla Salma, and scholarship of excellence from the National Center for Scientific and Technical Research in Morocco.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
MAB conceptualized the study, conducted the formal analysis and investigation, and wrote the original draft of the manuscript; MAB, AI, and MB developed the study methodology; MAB, AI, MB, and WL reviewed and edited the manuscript. MAB, AI, MB, NC, HA, LA, and TA helped in finding resources for the study; NC, AI, and MB supervised the study.