
Abstract
Background:
During leptospiral infection, the host innate immune response is initiated through recognition of pathogen-associated molecular patterns (PAMPs) by Toll-like receptor 2 (TLR2). Among these PAMPs, LipL32, Loa22, Lsa21, and lipopolysaccharide biosynthesis genes, are of particular interest.
Objective:
This study aimed to investigate these molecules’ genetic variability and evolutionary conservation in recently isolated clinical Leptospira strains from Sri Lanka.
Results:
We analyzed the whole-genome sequences of 25 clinical Leptospira isolates obtained from patients across Sri Lanka, sequenced using long-read technology and annotated using a standardized pipeline. Genes encoding LipL32, Loa22, Lsa21, and enzymes within the lipopolysaccharide biosynthesis locus were extracted and analyzed for phylogenetic relationships and sequence variation. LipL32 and Loa22 were highly conserved across all isolates, with only a single amino acid substitution observed in each. In contrast, genes associated with lipopolysaccharide biosynthesis, specifically those encoding glycosyl transferase and a sodium-dependent anion transporter, exhibited notable genetic variation, including multiple single nucleotide polymorphisms leading to amino acid changes. The Lsa21 gene was present only in Leptospira interrogans strains and showed no protein-level variation. Leptospira borgpetersenii isolates demonstrated strong conservation across all gene targets at both nucleotide and protein levels.
Conclusion:
Our findings highlight the high conservation of LipL32 and Loa22, reinforcing their potential as stable targets for molecular diagnostics and serological assays. In contrast, the variability observed in lipopolysaccharide biosynthesis genes suggests a possible role in immune evasion or adaptation, warranting further functional investigation. The restricted presence of Lsa21 in specific species also raises questions about its contribution to pathogenicity.
Keywords
Introduction
Leptospirosis, a globally prevalent infectious zoonosis, stems from a spiral-shaped bacterial species of the genus Leptospira.1,2 The disease is estimated to cause approximately 1.03 million infections and 58 900 deaths annually worldwide, 3 resulting in an annual productivity loss of approximately USD 29.3 billion. 4 Clinical manifestations range from mild febrile illness to severe multi-organ dysfunction 5 likely reflecting variation in both bacterial genomes and host immune responses. 6
Innate immune recognition of Leptospira is initiated by pattern recognition receptors, particularly Toll-like receptors (TLRs), which detect pathogen-associated molecular patterns (PAMPs) and trigger early inflammatory responses. 7 Among these, TLR2 has been implicated as a primary receptor for several leptospiral outer membrane components, including LipL32, Loa22, Lsa21, and lipopolysaccharide (LPS), each capable of inducing cytokine-mediated immune activation. 8
LipL32, the most abundant outer membrane lipoprotein in pathogenic Leptospira, is localized to the periplasmic leaflet, 9 may function as a calcium reservoir, 10 and activates TLR2 signaling. 11 Loa22, another conserved outer membrane protein with an OmpA domain, 12 similarly stimulates host responses via TLR2. 13 LPS, a major surface-exposed antigen composed of Lipid A, core oligosaccharide, and O-antigen, contributes to serovar specificity through genes within the rfb locus, 14 and also activates TLR2-dependent pathways.15-18 Lsa21, a leptospiral adhesin that binds extracellular matrix components, is another proposed TLR2-interacting protein. 19
Although the structural and immunostimulatory roles of key leptospiral PAMPs are well described, their genetic and protein-level variability across clinical isolates remains underexplored. Pathogenic Leptospira exhibit substantial genomic diversity, spanning Group 1 pathogens, Group 2 intermediates, and non-pathogenic saprophytes,20,21 which may influence host immune recognition and clinical outcomes. In particular, the extent of conservation and polymorphism in TLR2-interacting outer membrane components across species is poorly defined. To address this, we conducted a comparative genomic analysis of LipL32, Loa22, Lsa21, and LPS biosynthesis loci in 4 pathogenic species. This study aimed to elucidate molecular variation with relevance for innate immune activation and diagnostic development.
Methodology
Annotation of whole genome sequences of Leptospira spp.
A total of 25 Leptospira were successfully isolated from the blood cultures of acute undifferentiated febrile patients across 7 districts in Sri Lanka, representing diverse ecological and epidemiological settings between June 2016 and January 2019.22-25
For the present comparative genome analysis, whole-genome sequences (WGS) of these 25 isolates (designated with the prefix FMAS_) were analyzed. To facilitate comparative analysis of this study, 15 additional reference genomes were retrieved from the National Center for Biotechnology Information (NCBI) genome database (accessed 2024-04-09) (Table 1). These reference genomes included 4 NCBI-designated reference strains (3522CT, CUD06, FDAARGOS_203, and Piyasena), 2 Sri Lankan reference strains (6L-Int and 1L-Int), 1 intermediate pathogenic strain (ATCC BAA-1110), and 1 non-pathogenic saprophytic strain (Patoc 1). Additionally, 7 fully sequenced strains representing the 4 species identified among the Sri Lankan clinical isolates were included for comprehensive comparative analyses.
Sri Lankan Leptospira Isolates and Reference Genomes.

Bold sequences are tagged as “reference genomes” in NCBI database.
All 40 genome sequences were annotated simultaneously using Rapid Annotation using Subsystem Technology (RAST) (https://rast.nmpdr.org/) (annotation date: 2024-04-09). RAST was selected over other annotation platforms (such as NCBI’s own pipeline) because it allows for consistent, systematic annotation and direct comparison of all genomes within a unified analytical environment.
Gene and protein sequences specifically corresponding to leptospiral LipL32, Loa22, Lsa21, and the LPS O-antigen biosynthetic locus were separately downloaded from the NCBI nucleotide and protein databases (accessed between 2024-04-30 and 2024-05-20; Tables 2 and 3). From NCBI, we retrieved 1 complete LipL32 gene/protein sequence, 15 complete Loa22 gene/protein sequences, 12 partial Lsa21 sequences, and 1 complete LPS (O-antigen biosynthetic locus) gene sequence. The retrieved LPS gene sequence was translated into its corresponding protein sequence using the Expasy Translate Tool (https://web.expasy.org/translate/) to enable protein-level comparative analyses (accessed on 2024.05.15).
NCBI Available Gene Sequences for LipL32, Loa22, Lsa21, and LPS.

Bold sequences are the best matching sequences for all 40 whole genome sequences.
NCBI Available Protein Sequences for LipL32, Loa22, Lsa21, and LPS.

Bold sequences are the best matching sequences for all 40 whole genome sequences.
The best-matching protein sequences of LipL32, Loa22, Lsa21, and LPS O-antigen biosynthesis locus from NCBI were selected using the protein BLAST function within the RAST comparative tool, based on bit score and percent identity, for each of the 25 Sri Lankan isolates and the 15 reference genomes (Table 3). These matched protein sequences were then used as queries to extract corresponding gene and protein sequences from the annotated genomes using the RAST comparative protein BLAST tool. Two specific protein families encoded within the LPS biosynthesis locus glycosyltransferase (GT) and sodium-dependent anion transporter (SDAT) were extracted separately to facilitate more focused analyses. Nucleotide sequences of LipL32, Loa22, Lsa21, GT, and SDAT of 25 Leptospira isolates were submitted to GenBank via the BankIt submission portal (submitted on 2025.04.09, 2025.04.17, and 2025.04.18).
Phylogenetic Analysis
Multiple sequence alignments (MSA) of LipL32, Loa22, Lsa21, and LPS-related gene and protein sequences were performed using MUSCLE implemented in MEGA version 11 (MEGA 11). Phylogenetic relationships were reconstructed using the maximum likelihood (ML) method. Optimal substitution models for each gene/protein were determined separately using MEGA 11 prior to ML tree construction.
Previous studies indicated that the O-antigen region of LPS is associated with serogroup variability among Leptospira.14,26 However, initial analysis did not reveal substantial sequence variation within this region among our selected genomes. Therefore, phylogenetic analyses of the LPS locus were specifically conducted at the protein level for GT and SDAT proteins.
To evaluate phylogenetic reliability, bootstrap analysis was performed with 1000 replicates for LipL32, Loa22, and Lsa21. Due to computational considerations and initial findings, bootstrap analysis for GT and SDAT (LPS-associated proteins) was performed with 700 replicates. Phylogenetic trees were visualized and edited using FigTree v1.4.4 software (http://tree.bio.ed.ac.uk/software/figtree/).
Variation Analysis and IS Elements
Variation analyses were conducted using the Bacterial and Viral Bioinformatics Resource Center (BV-BRC) to identify single nucleotide polymorphisms (SNPs), insertions, and deletions within the target gene sequences. Species-specific SNP analyses were performed by referencing designated NCBI reference genomes: L. kirschneri serovar Cynopteri strain 3522CT, L. weilii strain CUD06, L. borgpetersenii serovar Ceylonica strain Piyasena, and L. interrogans serovar Copenhageni strain FDAARGOS_203. Additionally, insertion sequence (IS) detection was carried out for strains displaying amino acid substitutions using the IS Finder database (https://isfinder.biotoul.fr/) accessed on 2025-03-28.
Results
Gene and Protein Sequences of All 40 Leptospira Strains
Gene and protein sequences of LipL32, Loa22, Lsa21, and LPS O-antigen biosynthetic locus were successfully extracted from 25 Sri Lankan clinical isolates and 15 reference strains using best-matched NCBI protein sequences identified via BLAST analysis on the RAST comparative tool. Two distinct protein families, glycosyltransferase (GT) and sodium-dependent anion transporter (SDAT), were identified within the LPS gene locus and extracted separately. Sequence lengths are presented in Table 4, and BLAST metrics, including bit scores, percent identity, and gene locations, are provided in Supplementary information (Supplemental File 1). Gene sequences of LipL32, Loa22, Lsa21, GT, and SDAT of 25 Leptospira isolates obtained Accession numbers at Genbank https://www.ncbi.nlm.nih.gov/genbank/about/ and simultaneously make available to INSDC databases, European Nucleotide Archive (ENA) and DNA Data Bank of Japan (DDBJ).
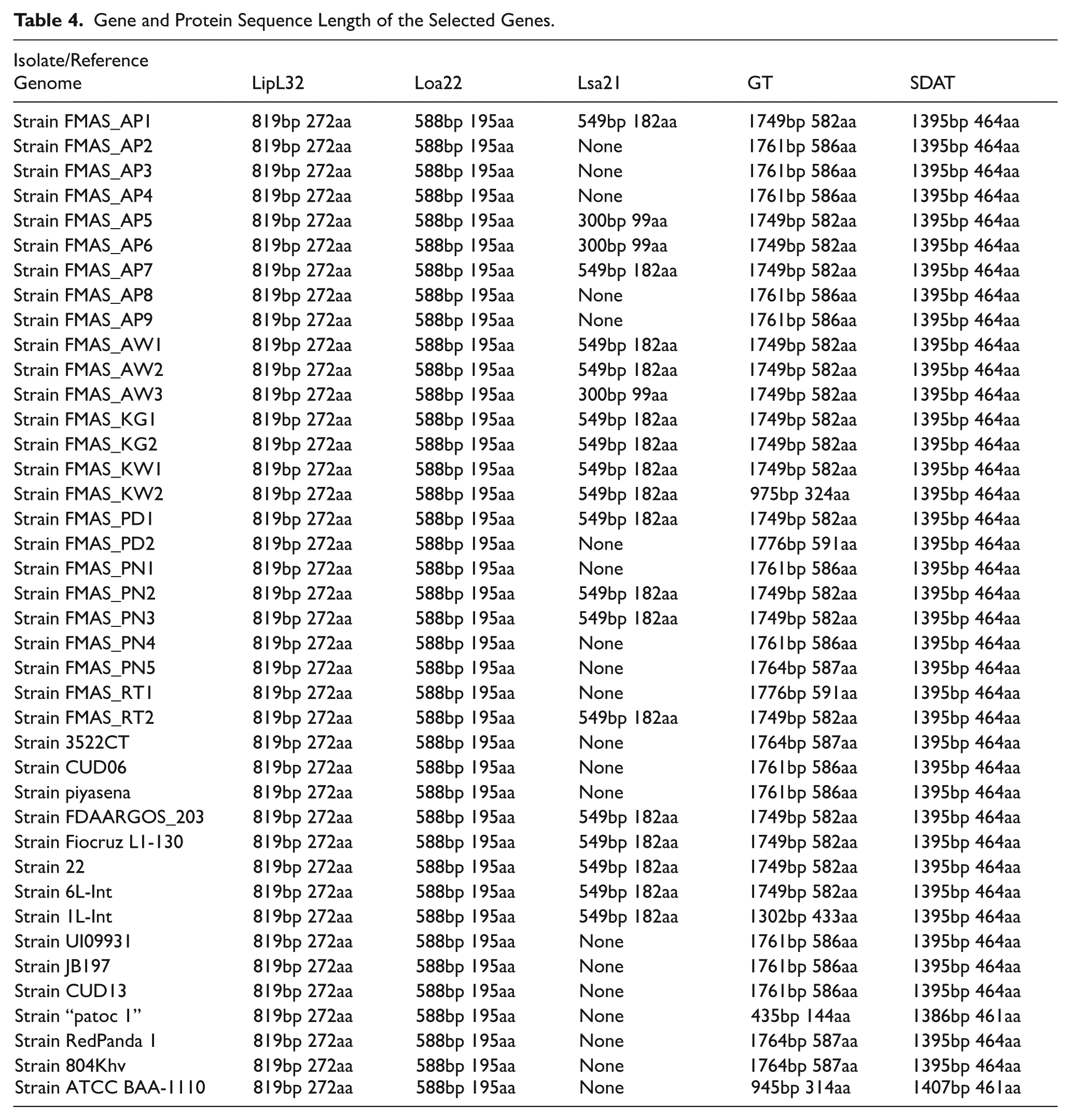
Gene and Protein Sequence Length of the Selected Genes.
Phylogenetic Analysis of the Leptospira Gene Sequences
We used the Maximum Likelihood method to infer the evolutionary history and an appropriate substitution model to regenerate the phylogenetic trees. Initial tree(s) for the heuristic search were obtained automatically by applying the Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated and then selecting the topology with the superior log likelihood value. The trees are drawn to scale, with branch lengths measured in the number of substitutions per site.
LipL32 Phylogenetic Analysis
The phylogenetic tree of LipL32 was regenerated using 40 of 819 bp gene sequences (Figure 1 shows the LipL32 original tree, and Figure S1 in Supplemental File 2 shows the LipL32 Bootstrapping tree). The phylogenetic tree revealed that strains are grouped into 4 clusters in 1 arm with distinct evolutionary relationships and Leptospira biflexa serovar Patoc strain “patoc 1” separately located as non-pathogenic saprophytes. 27 15 FMAS_ L. interrogans isolates and L. kirschneri were grouped in the same arm with 93% bootstrapping value together with NCBI reference genomes. FMAS_KG1 and FMAS_PN2 were closely clustered among all other L. interrogans. All the FMAS L. borgpetersenii strains were grouped in 1 cluster while the reference strain JB_197 shows a slight deviation. L. borgpetersenii clustering was supported by a 99% bootstrapping value. L. weilii were clustered with intermediate pathogenic L. licerasiae strain ATCC_BAA-1110 27 with 87% bootstrapping value. Within this cluster L. weilii were grouped with 100% bootstrapping support, while FMAS L. weilii strains FMAS_PD2 and FMAS_RT1 were clustered into 1 subgroup with 89% bootstrapping supportive value.

LipL32 Original tree. The evolutionary history was inferred by using the Maximum Likelihood method and Tamura 3-parameter model. The tree with the highest log likelihood (−3047.52) is shown. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Tamura 3 parameter model, and then selecting the topology with superior log likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. This analysis involved 40 nucleotide sequences. All positions with less than 95% site coverage were eliminated, that is, fewer than 5% alignment gaps, missing data, and ambiguous bases were allowed at any position (partial deletion option). There were a total of 819 positions in the final dataset. Evolutionary analyses were conducted in MEGA11.
Loa22 Phylogenetic Analysis
With a total of 588 bp nucleotides, all the strains regenerated the phylogenetic original tree (Figure 2) and the bootstrapping tree for Loa22 (Figure S2 in Supplemental File 2). L. biflexa serovar Patoc strain “patoc 1” and L. licerasiae strain ATCC_BAA-1110 were delineated separately with 100% bootstrapping value as non-pathogenic and intermediately pathogenic bacteria. Other pathogenic strains formed mainly 4 different clusters. 15 L. interrogans cluster sequences were sub grouped into 3 different clades with 99% bootstrap support. FMAS_PN5 L. kirschneri and reference strains were grouped in 1 cluster with a 93% bootstrap value. L. weilii and L. borgpetersenii were clustered into 1 arm with 100% bootstrapping subgrouping into 2 clades with 100% and 99% bootstrapping, respectively. All the L. borgpetersenii strains were clustered in 1 group except strain JB197 with a 99% bootstrap value.

Loa22 Original tree. The evolutionary history was inferred by using the Maximum Likelihood method and Tamura 3-parameter model. The tree with the highest log likelihood (−2345.79) is shown. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Tamura 3 parameter model, and then selecting the topology with superior log likelihood value. The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 25.94% sites). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. This analysis involved 40 nucleotide sequences. All positions with less than 95% site coverage were eliminated, that is, fewer than 5% alignment gaps, missing data, and ambiguous bases were allowed at any position (partial deletion option). There were a total of 588 positions in the final dataset. Evolutionary analyses were conducted in MEGA11.
Lsa21 Phylogenetic Analysis
We extracted Lsa21 gene sequence only from the L. interrogans strains and there were 2 different nucleotide lengths found as 549 and 300 bp. Phylogenetic tree was regenerated only using L. interrogans strains (Figure 3 shows the original tree and Figure S3 in Supplemental File 2 shows the bootstrapping tree). Other L. kirschneri, L. borgpetersenii, and L. weilii strains did not show matching Lsa21 gene sequences. FMAS_ L.interrogans were clustered into 2 evolutionary distinct clades with 88% and 67% bootstrapping values, while L. interrogans serovar Geyaweera was separately delineated.

Lsa21 Original tree. The evolutionary history was inferred by using the Maximum Likelihood method and Tamura 3-parameter model. The tree with the highest log likelihood (−427.93) is shown. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Tamura 3 parameter model, and then selecting the topology with superior log likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. This analysis involved 20 nucleotide sequences. All positions with less than 95% site coverage were eliminated, that is, fewer than 5% alignment gaps, missing data, and ambiguous bases were allowed at any position (partial deletion option). There were a total of 300 positions in the final dataset. Evolutionary analyses were conducted in MEGA11.
Glycosyl Transferase Phylogenetic Analysis
GT protein family was encoded from LPS gene sequence and we identified different nucleotide lengths as 1776, 1761, 1764, 1749, and 975 bp for different strains. Original tree and the bootstrapping tree were regenerated using those gene sequences (Figure 4 shows the original tree and Figure S4 in Supplemental File 2 shows the bootstrapping tree). All L. interrogans except FMAS_KW2 and L. kirschneri were grouped into 1 cluster with evolutionary distances. L. biflexa serovar Patoc strain “patoc 1” and L. licerasiae strain ATCC_BAA-1110 were located separately but closer to L. interrogans and L. kirschneri cluster. L. weilii and L. borgpetersenii were grouped into 1 arm and then were sub grouped into 2 clades. FMAS_ L. borgpetersenii were clustered together without evolutionary differences, except the reference strain L. borgpetersenii strain JB_197. FMAS_ L. weilii were clustered together without evolutionary distances with bootstrap supporting value of 100%. Phylogenetic original tree and bootstrapping tree clearly evidenced the variations among GT gene sequences of the strains even within 1 species of L. interrogans, L. kirschneri and L. weilii.

Glycosyl transferase Original tree. The evolutionary history was inferred by using the Maximum Likelihood method and Tamura 3-parameter model. The tree with the highest log likelihood (−7977.77) is shown. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Tamura 3 parameter model, and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 4.6921)). The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 10.81% sites). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site (next to the branches). This analysis involved 40 nucleotide sequences. All positions with less than 95% site coverage were eliminated, that is, fewer than 5% alignment gaps, missing data, and ambiguous bases were allowed at any position (partial deletion option). There were a total of 1189 positions in the final dataset. Evolutionary analyses were conducted in MEGA11.
Sodium Dependent Anion Transporter Phylogenetic Analysis
We identified the gene sequence length is as 1395 bp for each strain except L. licerasiae strain ATCC_BAA-1110 (1407 bp) and regenerated the original tree and bootstrapping tree (Figure 5 shows original tree and Figure S5 in Supplemental File 2 shows bootstrapping tree). Intermediate pathogenic L. licerasiae strain ATCC_BAA-1110 and non-pathogenic strain L. biflexa serovar Patoc strain “patoc 1” were located separately. Other strains were clustered into 4 main groups. L. interrogans strains were grouped in 1 cluster, supporting 99% bootstrapping value with minimum evolutionary distances. L. kirschneri strains were clustered into 1 clade with 99% bootstrapping value and FMAS_PN5 located with evolutionary distance from other reference strains. L. weilii strains were clustered with 100% bootstrapping value, and those of FMAS and reference strains were showed minimum evolutionary distance. As similar as in other gene sequences, L. borgpetersenii strains were grouped in 1 cluster, supporting 100% bootstrapping value with a minimum evolutionary distance for L. borgpetersenii strain JB_197.
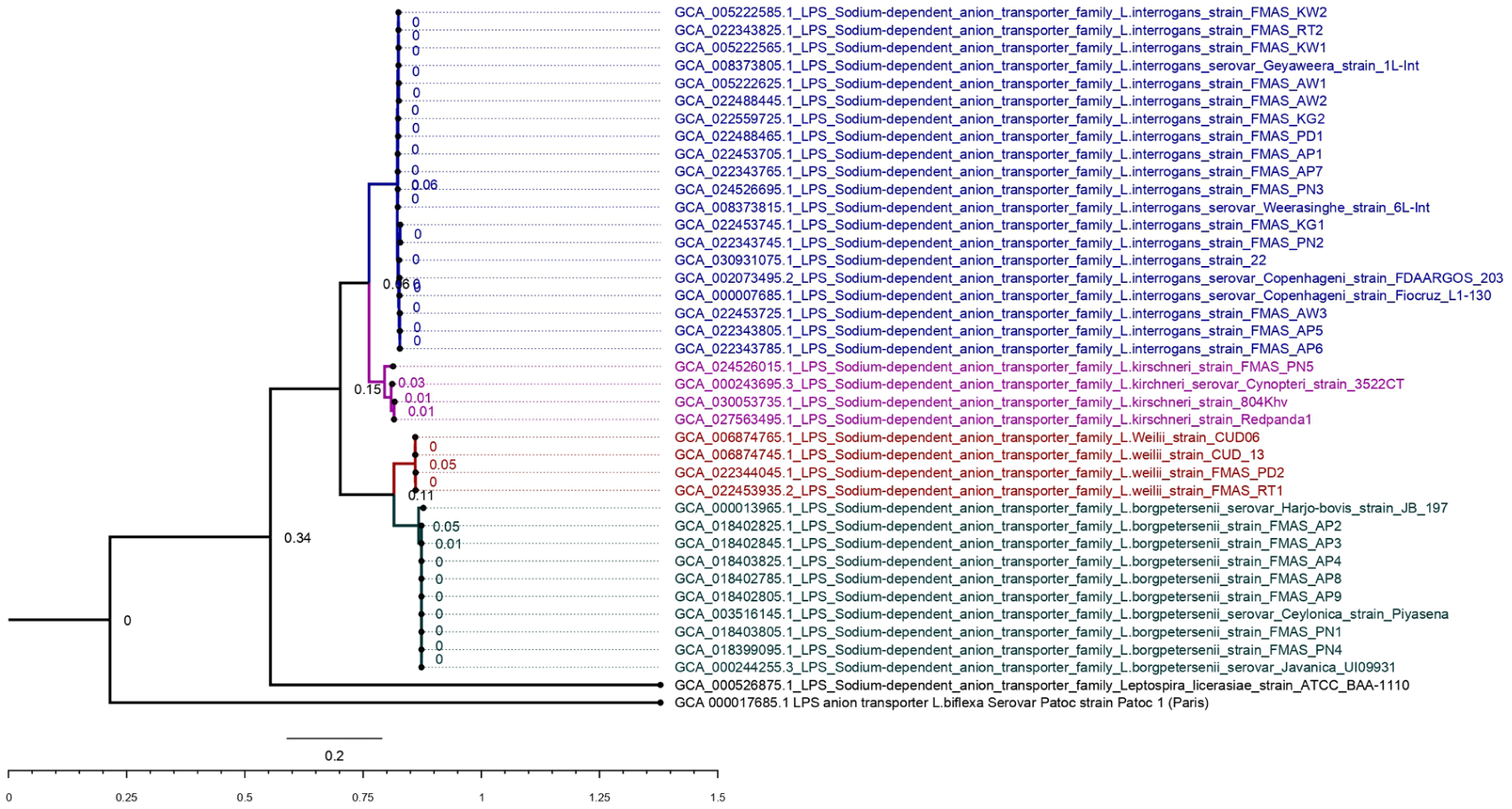
Sodium dependent anion transporter Original tree. The evolutionary history was inferred by using the Maximum Likelihood method and Tamura-Nei model. The tree with the highest log likelihood (−6865.69) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Tamura-Nei model, and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 2.6291)). The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 18.03% sites). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. This analysis involved 40 nucleotide sequences. All positions with less than 95% site coverage were eliminated, that is, fewer than 5% alignment gaps, missing data, and ambiguous bases were allowed at any position (partial deletion option). There were a total of 1395 positions in the final dataset. Evolutionary analyses were conducted in MEGA11.
Phylogenetic Analysis of the Leptospira protein sequences to determine the evolutionary relatedness (GT and SDAT)
GT and SDAT protein sequences of 20 L. interrogans strains belong to 7 serogroups were regenerated the ML trees (Figures 6 and 7). All the strains belong to Autumnalis, Pyrogenes, Icterohaemorrhagiae, Bataviae and Weerasinghe were grouped into 1 cluster with less divergence, while serovar Geyaweera was outgrouped. SDAT protein sequences were also clustered in 1 group representing serogroup Autumnalis, Pyrogenes, Weerasinghe, and Geyaweera. Serogroup Icterohaemorrhagiae and Bataviae were clustered together, while Serogroup strains FMAS_AP5, AP6, AW3 were grouped in 1 cluster. Both protein sequences showed close evolutionary relationships for all strains with less divergence that did not demonstrate serogroup specific sequences within L. interrogans species.

Glycosyl Transferase Original tree-protein. The evolutionary history was inferred by using the Maximum Likelihood method and General Reversible Chloroplast model. The tree with the highest log likelihood (−3466.03) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the JTT model, and then selecting the topology with superior log likelihood value. The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 7.54% sites). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site (next to the branches). This analysis involved 20 amino acid sequences. All positions with less than 95% site coverage were eliminated, that is, fewer than 5% alignment gaps, missing data, and ambiguous bases were allowed at any position (partial deletion option). There were a total of 484 positions in the final dataset. Evolutionary analyses were conducted in MEGA11.

Sodium dependent anion transporter original tree-protein. The evolutionary history was inferred by using the Maximum Likelihood method and General Reversible Mitochondrial model. The tree with the highest log likelihood (−1356.16) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the JTT model, and then selecting the topology with superior log likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site (next to the branches). This analysis involved 20 amino acid sequences. All positions with less than 95% site coverage were eliminated, that is, fewer than 5% alignment gaps, missing data, and ambiguous bases were allowed at any position (partial deletion option). There were a total of 464 positions in the final dataset. Evolutionary analyses were conducted in MEGA11.
Multiple Sequence Alignment and Single Nucleotide Polymorphism
Different gene and protein polymorphisms were found among the FMAS strains during the MSA and SNP analysis (Table 5 and Supplemental File 3). We observed greater sequence dissimilarities in GT and SDAT which were extracted from LPS O-antigen biosynthetic locus among all strains except L. borgpetersenii. Further LipL32 is highly conserved among all the sequences than Loa22. There are no protein polymorphisms for Lsa21 which was a moderate conserved region which showed a few gene SNPs.
Number of Gene/Protein SNPs of Each FMAS Strain.

LipL32 Variation Analysis
Gene sequences of 819 bp and protein sequences of 272 aa were subjected to perform MSA using Mafft and SNP on BV-BRC 3.42.3. All the FMAS L. interrogans isolates located in 1 branch sharing similar nucleotide sequences with reference to L. interrogans serovar Copenhageni strain FDAARGOS_203, except strain FMAS_KG1 and FMAS_PN2 which show close phylogenetic relationships with SNPs (Table 6 and Figures S6-S8 in Supplemental File 2). None of these SNPs have led to protein polymorphisms. Gene and protein sequences of FMAS L. borgpetersenii strains were located in 1 branch sharing similar sequences with reference to L. borgpetersenii serovar Ceylonica strain Piyasena. Variation analysis revealed that FMAS L. borgpetersenii strains do not exhibit any gene or protein polymorphism (Table 6 and Figures S9 and S10 in Supplemental File 2). L. kirschneri FMAS_PN5 showed 3 SNPs, without amino acid sequence differences (Figures S11-S14 in Supplemental File 2). Two SNPs of L. weilii FMAS_PD2 and L. weilii FMAS_RT1 showed protein polymorphism, as evidenced in the phylogenetic tree with greater sequence divergence compared to other FAMS strains (Table 6 and Figure 8).
LipL32 Gene and Protein Polymorphisms.


SNPs of FMAS L. weilii LipL32. (A) SNP (T480C), (B) SNP (A548G,T552C), and (C) SNP (Lys183Arg).
Loa22 Variation Analysis
Loa22 gene and protein sequences (588 bp and 195 bp) were subjected to MSA and SNP analysis using Mafft and SNP on BV-BRC 3.42.3 (Table 7). FMAS_ L. interrogans showed 3 different gene SNPs (Figures S15-S18 in Supplemental File 2), which haven’t led to protein polymorphism. Phylogenetic relationship revealed the highest sequence divergence of FMAS L. borgpetersenii among other strains, while there were no Loa22 gene or protein polymorphisms with reference to L. borgpetersenii serovar Ceylonica strain Piyasena (Figure S19 in Supplemental File 2). FMAS_L. weilii showed Loa22 gene polymorphism that has not led to protein polymorphism with less sequence divergence than L. borgpetersenii (Figures S20 and S21 in Supplemental File 2). L. kirschneri strain FMAS_PN5 which is closely related to L. interrogans showed 5 different gene polymorphisms with 1 protein polymorphism Lys to Arg at 180 aa (Figure 9).
Loa22 Gene and Protein Polymorphisms.


SNPs of FMAS_L. kirschneri Loa22. (A) (T390C), (B) (T510A, G528A), (C). (C543T, T558C), and (D).(Lys180 Arg).
Lsa21 Variation Analysis
We found Lsa21 gene sequence only in FMAS L. interrogans strains and 2 lengths of gene sequences were identified as 549 bp (182aa) and 300 bp (99aa). MSA and SNP analysis evidenced that, though there were SNPs among the gene sequences, no protein polymorphisms were found (Table 8 and Figures S22 and S23 in Supplemental File 2). FMAS_AP1, AP7, AW1, AW2, KW1, KW2, PN3, RT2, PN2 showed gene SNPs which show greater sequence divergence from other L. interrogans strains.
Lsa21 Gene and Protein Polymorphisms.

Variation Analysis of Glycosyl Transferase
Owing to the gene and protein sequence differences of GT, several gene and protein polymorphisms were identified during MSA and SNP analysis (Table 9). Other than the SNPs, there were several deletions found in the FMAS_ L. interrogans. FMAS_KW2 was delineated separately from other L. interrogans due to its nucleotide deletions and SNPs. Separate delineation of FMAS_KW2 from other L. interrogans in the MSA tree is supported by the high sequence divergence reported in the phylogenetic tree. Five different protein polymorphisms were shown by other FMAS_ L. interrogans strains (Table 9 and Figures S24-S27 in Supplemental File 2). Further we observed gene polymorphisms leading to protein polymorphisms in both L. kirschneri and L. weilii FMAS_ strains (Table 9 and Figures S28-S33 in Supplemental File 2). As observed in the LipL32, Loa22, and Lsa21 analysis, L. borgpetersenii strains did not show any gene or protein polymorphisms (Figure S34 in Supplemental File 2).
Glycosyl Transferase Gene and Protein Polymorphisms.

Variation Analysis of Sodium dependent anion Transporter
Gene and protein sequences with 1395 bp and 464aa were subjected to MSA and SNP analysis. FMAS_strains of L. interrogans and L. kirschneri showed gene and protein polymorphisms for SDAT (Table 10 and Figures S35-S40, S42-S46 in Supplemental File 2). Evolutionary wise closely related L. interrogans and L. kirschneri were located with close proximity in the phylogenetic tree. In line with previous sequences, L. borgpetersenii did not show any gene or protein polymorphisms for Sodium dependent anion transporter (Figure S41 in Supplemental File 2). Although we observed gene SNP of FMAS_L. weilii strain for SDAT, it has not led to protein polymorphism (Table 10 and Figures S47-S49 in Supplemental File 2).
Sodium Dependent Anion Transporter Gene and Protein Polymorphisms.

IS elements of Glycosyl Transferase and Sodium dependent anion transporter We found different IS elements in GT and SDAT gene sequences (Tables 11 and 12).
IS Elements of Glycosyl Transferase.

IS Elements of Sodium Dependent Anion Transporter.

Discussion
This study provides insights into the evolutionary relationships and genetic polymorphisms of key leptospiral pathogen-associated molecular patterns (PAMPs), LipL32, Loa22, Lsa21, glycosyltransferase (GT), and sodium-dependent anion transporter (SDAT) across 25 recently isolated Sri Lankan clinical isolates of Leptospira. 25 Our analyses confirmed that these gene and protein sequences are highly conserved within pathogenic species but show significant divergence from intermediate and non-pathogenic Leptospira, reflecting their importance in pathogenesis and host adaptation.
Within the Sri Lankan pathogenic set, LipL32 was highly conserved at the amino-acid level, with pairwise comparisons showing negligible non-synonymous change. Phylogenetic reconstruction separated pathogenic from intermediate/saprophytic taxa as expected. These findings support LipL32 as a stable TLR2-interacting lipoprotein and a robust diagnostic antigen within local pathogenic Leptospira. Loa22 displayed strong species-level structure with minimal intra-species variation. All L. interrogans isolates formed a single lineage with no amino-acid substitutions detected across the local set. Conservation of this OmpA-like, virulence-associated protein is consistent with preserved TLR2-recognition surfaces across circulating L. interrogans. Lsa21 was detected exclusively in L. interrogans among the analyzed isolates and occurred in 2 length variants (a longer, full-length form and a shorter, truncated form). Identity was high within each length class but substantially lower between classes, indicating species restriction with within-species structural variability that could modulate TLR2 engagement. The absence of Lsa21 from non L. interrogans species implies that this antigen cannot account for TLR2-mediated responses in those infections. For gt and sdat, sequence comparisons and phylogeny showed clear species-level differentiation with limited diversity within species. A common gt length was observed in L. interrogans, whereas shorter gt variants occurred in L. borgpetersenii; sdat exhibited a similar species-stratified pattern. Serovar-level resolution was not achieved using gt/sdat alone, and only partial clustering by serogroup was evident, indicating that full O-antigen locus analyses would be required for serogroup/serovar inference. One L. borgpetersenii strain (JB197; serogroup Sejroe) was more divergent within its clade, consistent with host-origin differences noted in the results.
Most studies on LipL32 phylogeny have identified LipL32 as a highly conserved, immunogenic, outer membrane protein coding gene sequence among pathogenic Leptospira spp9,28-32 in consistent with our findings. A bioinformatics approach to 3D modeling of pathogenic Leptospira LipL32 showed limited diversity in terms of pairwise alignment, hydrophobic group, hydrophilic group, and number of turns, among the strains of L. interrogans, L. borgpetersenii, L. santarosai and L. alstoni. 33 Aligned with our findings comparative genome studies on Loa22, demonstrated that Loa22 is also conserved among the pathogenic Leptospira serovars.34,35 Well conserved gene/protein sequences can be utilized, as PCR targets and ELISA antigens, while Polymerase Chain Reaction (PCR) and Enzyme-Linked Immunosorbant Assay (ELISA) are being 2 common diagnostic tools of Leptospirosis. 36 As of now 16S rRNA, LipL32 37 are widely used target sequences for PCR and Loa22 can also be used. One research group studied the effect of substitution of Lysine to Arginine on protein stability and structure, that we observed in both LipL32 and Loa22 of FMAS L. weilii and L. kirschneri strains respectively. It suggested the increase of protein stability and unfavorable protein folding due to the presence of Arginine over Lysine with Guanodino group to Amine group that particularly affect the protein interaction and binding affinity. 38 Mutation properties could enhance the interaction between LipL32, Loa22, and TLR2 to develop human immune responses and this may associate with pathogenesis of Leptospira spp. Arginine to Lysine substitution in FMAS strains were observed with reference to the reference genomes in NCBI, L. weilii strain CUD06 and L. kirschneri serovar Cynopteri strain 3522CT which were isolated from Gray wolves urine and bats respectively. Taking source of Leptospira origin and mutations into account, these findings emphasize the need of further structural based functional analysis to investigate whether the pathogenesis is affected by the mutations. Other studies also showed the increase of enzyme activity, 39 enhance of antimicrobial activity of peptides, 40 increase of cellular uptake of chlorotoxin 41 due to the substitution of Arginine over Lysine. Similarly, it is important to investigate whether this Arginine over Lysine substitution influences the pathogenesis of Leptospira.
Although there are evidence to report the presence of Lsa21 in L. interrogans strains and the absence in saprophytes,19,42 no studies explicit mention the presence of Lsa21 in other Leptospira species. Nevertheless there are two L. kirschneri Lsa21 partial gene sequences directly submitted to NCBI (Accession: OQ450472.1 and Accession: OQ941635.1), without published data in literature. Further there were no good hits/best matching sequences related with those Lsa21 in our FMAS strains or other reference genomes that we used. It appears that Lsa21 is mainly express in L. interrogans and we did not observe Lsa21 in other species of our analysis. With further confirmation of the presence of Lsa21 among species of Leptospira, would help to differentiate the L. interrogans from other species and enhance the understanding of the pathogenesis of Leptospira spp. specifically associate with Lsa21.
Eventhough the O-antigen is an epitope for serogroup specificity,26,43 in our analysis we found that the GT and SDAT have clustered/differentiated the strains into 4 species, but not into serogroups/serovars. One research group from China has developed a PCR base O-antigen genotyping method using 6 epidemic serogroups; Canicola, Autumnalis, Grippotyphosa, Hebdomadis, Icterohaemorrhagiae, and Sejroe. 44 Nevertheless our finding demonstrated that GT and SDAT can be utilized to differentiate the serogroups/serovars in different species, but not in same species. Although there were no comprehensive studies on amino acid substitutions observed in those 2 proteins, it is worth to suggest the need of further studies on amino acid substitutions presented in our analysis, to determine whether the changes contribute to disease pathogenesis. Further, we found IS elements in GT and SDTA, that could be a main reason for the presence of SNPs and protein substitutions that may lead to disease pathogenesis. 45 Some of the amino acid substitutions presented in GT, Glutamic acid to Lysine at codon 332 caused adult onset Alexander disease, 46 Glutamic acid 192 Glutamine restrict the specificity of Thrombin 47 were reported. Lysine to Glutamic acid substitution in GT is also an impactful, since the charge changes of amino acids may potentially disrupt substrate binding and alter the enzyme function. 48 Arginine to Glutamic acid substitution in SDAT may disrupt electrostatic interactions in critical substrate binding regions due to charge changes of amino acids. 49 Those research groups have demonstrated the mutation results according to the changes of amino acid properties as polarity, size and shape, acidic-basic, chemical reactivity that decide the protein structure, stability, function, and interactions. Likewise, the understanding of mutation properties of GT and SDAT in relation to leptospirosis is crucial and need to be clarified in future research efforts.
One major strength of our study is the use of whole genome sequencing data from low-passage culture isolates obtained from patients’ blood. Hence, we were able to generate reliable data with limited or no mutations due to low passage. Further, we compared LipL32, Loa22, Lsa21, Glycosyl transferase and Sodium dependent anion transporter genes that are more immunogenic among 25 Leptospira strains from Sri Lanka, which may be useful for researchers who are analyzing data to understand leptospirosis pathogenesis. One caveat of this study is that we were unable to perform the 3D modeling of the proteins due to time constraints. Another limitation of the study is the absence of a formal sample size calculation, which may affect the generalizability of the findings.
Conclusions
This analysis identifies that LipL32 and Loa22 are well conserved gene regions while O-antigen biosynthetic genes GT and SDAT presented a number of amino acid substitutions among 25 FMAS Leptospira strains. These findings offer valuable insights for the development of diagnostic tools and a deeper understanding of disease pathogenesis. As diagnostic tools, LipL32 and Loa22 can be utilized as PCR target sequences and ELISA antigens as the well conserved regions among pathogenic Leptospira. SNPs followed by amino acid substitutions may lead to structural and functional differences of LipL32, Loa22 and O-antigen region that can associate with disease pathogenesis and strengthening the host-pathogen interaction. Besides, Lsa21 is unique to L. interrogans as it is not present in L. kirschneri, L. weilii or L. borgpetersenii strains. Further confirmation of Lsa21 expression in Leptospira, would be helpful in species differentiation and understanding of Lsa21 gene related disease pathogenesis.
Supplemental Material
sj-pdf-1-evb-10.1177_11769343251389782 – Supplemental material for Comparative Genome Analysis of 25 Sri Lankan Leptospira Isolates Outer Membrane Receptors That Interact With Human TLR2
Supplemental material, sj-pdf-1-evb-10.1177_11769343251389782 for Comparative Genome Analysis of 25 Sri Lankan Leptospira Isolates Outer Membrane Receptors That Interact With Human TLR2 by Chamila Kappagoda, Indika Senavirathna, Dinesha Jayasundara, Janith Warnasekara, Thilini Agampodi and Suneth Agampodi in Evolutionary Bioinformatics
Supplemental Material
sj-xlsx-2-evb-10.1177_11769343251389782 – Supplemental material for Comparative Genome Analysis of 25 Sri Lankan Leptospira Isolates Outer Membrane Receptors That Interact With Human TLR2
Supplemental material, sj-xlsx-2-evb-10.1177_11769343251389782 for Comparative Genome Analysis of 25 Sri Lankan Leptospira Isolates Outer Membrane Receptors That Interact With Human TLR2 by Chamila Kappagoda, Indika Senavirathna, Dinesha Jayasundara, Janith Warnasekara, Thilini Agampodi and Suneth Agampodi in Evolutionary Bioinformatics
Supplemental Material
sj-xlsx-3-evb-10.1177_11769343251389782 – Supplemental material for Comparative Genome Analysis of 25 Sri Lankan Leptospira Isolates Outer Membrane Receptors That Interact With Human TLR2
Supplemental material, sj-xlsx-3-evb-10.1177_11769343251389782 for Comparative Genome Analysis of 25 Sri Lankan Leptospira Isolates Outer Membrane Receptors That Interact With Human TLR2 by Chamila Kappagoda, Indika Senavirathna, Dinesha Jayasundara, Janith Warnasekara, Thilini Agampodi and Suneth Agampodi in Evolutionary Bioinformatics
Footnotes
ORCID iDs
Author Contributions
Chamila Kappagoda: Conceptualization, Methodology, Software, Formal analysis, Writing—Original Draft, Visualization, Investigation. Indika Senavirathna: Conceptualization, Methodology, Software, Writing—Review and Editing, Supervision. Dinesha Jayasundara: Resources, Writing—Review and Editing. Janith Warnasekara: Writing—Review and Editing. Thilini Agampodi: Writing—Review and Editing. Suneth Agampodi: Conceptualization, Methodology, Software, Writing—Review and Editing, Supervision.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The gene sequences generated during the current study are available in the GenBank repository with accession numbers of; GenBank: PV471625, GenBank: PV539477- PV539545, GenBank: PV551211- PV551255. The data are simultaneously available to INSDC, ENA, and DDBJ. Other data sets are included within the article and its supplementary files.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.