
Abstract
Background:
Statistical methods developed to address various questions in single-cell datasets show increased variability to different parameter regimes. In order to delineate further the robustness of commonly utilized methods for single-cell RNA-Seq, we aimed to comprehensively review scRNA-Seq analysis workflows in the setting of dimension reduction, clustering, and trajectory inference.
Methods:
We utilized datasets with temporal single-cell transcriptomics profiles from public repositories. Combining multiple methods at each level of the workflow, we have performed over 6k analysis and evaluated the results of clustering and pseudotime estimation using adjusted rand index and rank correlation metrics. We have further integrated neural network methods to assess whether models with increased complexity can show increased bias/variance trade-off.
Results:
Combinatorial workflows showed that utilizing non-linear dimension reduction techniques such as t-SNE and UMAP are sensitive to initial preprocessing steps hence clustering results on dimension reduced space of single-cell datasets should be utilized carefully. Similarly, pseudotime estimation methods that depend on previous non-linear dimension reduction steps can result in highly variable trajectories. In contrast, methods that avoid non-linearity such as WOT can result in repeatable inferences of temporal gene expression dynamics. Furthermore, imputation methods do not improve clustering or trajectory inference results substantially in terms of repeatability. In contrast, the selection of the normalization method shows an increased effect on downstream analysis where ScTransform reduces variability overall.
Background
Intra-tumor heterogeneity has recently become a central focus of cancer research secondary to the limited response of patients to targeted therapies. These failures are driven by Darwinian evolution, by heritable variation and selection through time. One source for the subsequent intra-tumor heterogeneity is the variation driven by stochasticity in transcriptional activity modulated by epigenetic processes.1,2 This change in overall composition is further modulated by the selective advantage of pre-existing resistant cells or clonal expansion of drug-tolerant cells mediated by complex interactions between cells and the microenvironment.3-6 Although previous efforts have made significant progress in understanding the complex cancer dynamics using bulk sequencing data, single-cell sequencing methods have allowed for novel insights by probing this heterogeneity directly—including during temporally varying processes. For instance, Lee et al 7 identified transcriptional heterogeneity as one of the key factors for promoting the clonal expansion of drug-tolerant sub-population leading to the evolution of resistance. Similarly, Kim et al 8 identified distinct sub-populations resistant to treatment in lung adenocarcinoma patients using single-cell RNA-Seq, Furthermore, relatively recently, single-cell sequencing coupled with mathematical models allowed for investigation of Darwinian dynamics, specifically treatment-induced selection pressure and transcriptional stochasticity at the single-cell level.9-11
Investigating transcriptional regulation, single-cell sequencing also paved the way for pseudotime/trajectory estimation (PTE) to delineate temporal dynamics during differentiation and resistance evolution. Specifically, PTE aims to find low dimensional proxy for the underlying transcriptional activity accounting for the temporal information. However, due to the stochasticity inherent in evolution, PTE poses additional challenges where replicate experiments can show divergent dynamics leading to the evolution of distinct resistance mechanisms.12,13 For instance, during multipotent progenitor trophoblast differentiation, stages of organization (endpoints) are clearly defined based on morphological characteristics hence we can reliably deduce functional mechanisms through time. 14 However, as we show in detail below, using the same analysis methods with slight differences in pre-processing parameters (number of genes expressed), can result in very different PTE orderings of cells in the setting of the evolution of resistance leading to increased diversity of identified mechanisms. Analysis of single-cell data is further complicated by the technical noise in library preparation strategies due to capture efficiencies at both cell (empty/multi-cell droplets) and transcript level.
In order to alleviate some of the issues with single-cell analysis, various analysis methods aim for robust imputation, outlier detection, and quantification of gene expression. For instance, previous studies utilized imputation methods to reduce the effects of zero-counts due to dropouts in RNA-Seq datasets.15,16 In addition to individual methods, multiple packages integrate different analysis stages and tools in unified frameworks; Seurat,17-20 SCANPY, 21 BUStools.22,23 However, the increased number of available tools, and continued proliferation of them also requires careful selection of methods and associated parameters which can result in significant differences. This issue has been partially addressed before. Specifically, 2 comprehensive combinatorial evaluation studies have been conducted in order to evaluate different analysis workflows.24,25 Tian et al 25 using cell-mixture experiments showed relatively good correlations between ground-truth and estimated trajectories using Slingshot or DDRTree. Similarly, Saelens et al 24 showed improved performance for these methods using topological similarity metrics. While illuminating, a major limitation of these studies is that the methods are applied on non-cancer (embryonic differentiation) processes or cancer cells in relatively homogeneous settings (without selection pressure). For instance, mixture experiments conducted by Tian et al are limited to linear trajectories. In contrast, evolution under selection pressure can result in increased variability and non-linear patterns of transcriptional change.26-28 As most tumors do not grow in these conditions, it is crucial to evaluate the available methods under selection pressure with temporal information as well. For this purpose, in this manuscript, we report a benchmarking study in which we evaluate the available methods in a combinatorial fashion similar to Tian et al and Saelens et al focusing on the repeatability of PTEs. We hope that by evaluating the scRNA-Seq methods rigorously for settings applicable to the evolution of resistance in cancer, we will enable more robust and reproducible application of single-cell sequencing technologies and experimental designs for future studies.
Methods
Single-cell RNA-Seq (scRNA-Seq) analysis follows similar strategies with bulk RNA-Seq where pre-processing is followed by normalization for library size and downstream analysis (see Figure 1 for a schema of a typical workflow). Due to the large number of cells being captured non-linear dimension reduction techniques have been extensively used for clustering and trajectory identification such as t-SNE and UMAP.29,30 In addition to dimension reduction methods, scRNA-Seq datasets can be zero-inflated due to increased technical noise, hence various imputation approaches have been proposed. Furthermore, a general trend in the scRNA-Seq analysis is to filter out genes that show relatively low variation across the dataset and filter out cells that express a low number of genes. Although this is a valid strategy similar to bulk RNA-Seq analysis, the cutoff for the number of top varying genes to select and the number of genes expressed are generally arbitrary chosen, hence we aim to evaluate the effects of filtering genes and cells based on different thresholds as well. For this purpose, we combine various methods for different levels of analysis in a combinatorial fashion and evaluate identified trajectories in terms of cell orderings (Also note that combinatorial workflows are represented by small icons in downstream figures as column and row labels). Furthermore, since the ground-truth trajectories do not necessarily associate linearly with time in heterogeneous processes (eg, drug resistance), we have profiled clustering performance as well. 28 (See Appendix for a detailed description of methods and parameters.)

Schematic of general analysis steps and methods used for combinatorial workflows. Quality filtered raw read counts are processed through a step to reduce possible zero-count inflation by 1 of 2 imputation methods: ScImpute, DrImpute (or no imputation). Preprocessed data is normalized by 3 methods; ScTransform, Deconvolution, and DCA followed by dimension reduction using 5 methods; UMAP, UMAP + PAGA, t-SNE, VAE, DM. Finally, 1 of 4 trajectory inference methods is used; Slingshot, DDRtree, and WOT. Overall, we have utilized 6144 analyses for PTE including the data subsets. (Note that the icons representative of individual methods are used to ease the interpretability of combinatorial workflows in downstream figures. Created with BioRender.com.)
We have utilized both publicly available datasets and a previously generated in-house dataset with variable number of cells, depth, and complexity of the underlying process (Table 1). TKI Treatment dataset was previously generated to investigate transcriptional dynamics of resistance evolution to 3 Tyrosine kinase inhibitors (TKIs); Alectinib, Lorlatinib, and Crizotinib in lung cancer. To generate scRNA-Seq data with temporal information, cells were sampled at 4 hours (Alectinib only), 48 hours, 3 w, and 20 to 24 w and sequenced. As we have hypothesized, this dataset represents a biologically heterogeneous example of an evolutionary process hence crucial to evaluate PTEs. The Pancreatic cell maturation dataset contains transcriptional profiles of α and β cells during differentiation process at 7 time-points: embryonic day 17.5 and postnatal days 0, 3, 9, 15, 18, and 60 representing a relatively more homogeneous process with roughly linear sampling times. Neurodegeneration dataset is generated to investigate the transcriptional dynamics of microglial cells isolated from Hippocampus at weeks 0, 1, 2, and 6 in CK-p25 inducible mouse model. E2 Treatment temporal scRNA-Seq is performed on 2 cell lines (MCF7, T47D) during 17β-estradiol (E2) treatment at 0, 3, and 6, and 12 hours to investigate temporal transcriptional dynamics of estrogen associated pathways in breast cancer. This dataset, however, contains the least number of captured cells sequenced at relatively higher depth.
Datasets utilized in the study where the number of cells and genes are given prior to subset generation after quality control.

Each dataset is preprocessed with different gene- and cell-level quality thresholds to generate 12 subsets and the overlap in estimated trajectories are quantified using rank correlation. We have focused on the repeatability of identified PTEs and aimed to use methods/strategies widely adopted in the community. Additionally, we utilize a neural network approach for dimension reduction to evaluate whether more complex models show any advantage when high-throughput single-cell datasets are used. Since neural networks have been extensively utilized for wide variety of problems in the form of autoencoders 35 and relatively recently stochastic alternatives have been used for—omics datasets as well,36-39 neural networks naturally lend themselves to the analysis of single-cell datasets. For comparison of the effect of imputation, we have used ScImpute, DrImpute which showed improved performance in various datasets and Deep Count Autoencoder (DCA) an autoencoder model aiming to combine de-noising and imputation in a single step.15,16,40 We use 2 methods for normalization: Deconvolution and ScTransform.41,42 For DCA, since gene-wise dispersion and mean parameters are already estimated, we only used library size normalization. As scRNA-Seq clustering is an important step utilized in the analysis of various datasets, we wanted to evaluate how robust the clustering results are when different workflows are used as well. For this purpose, we coupled the Leiden clustering with 5 dimension reduction techniques; UMAP, PAGA + UMAP, t-SNE, VAE, and Diffusion Maps (DM) and evaluated the overlap of clusters using adjusted rand index (ARI).29,30,43-46 We have additionally included TooManyCells for clustering, however, due to hardware limitations we used only the Pancreatic Maturation, Neurodegeneration datasets. Furthermore, E2 Treatment dataset resulted in a single cluster across different workflows and subsets possibly due to the low number of cells hence results are not shown. 47 For trajectory inference, we evaluated 4 methods commonly used in scRNA-Seq; Slingshot, Palantir, DDRTree, and WOT.48-51 However, Slingshot operates on dimension reduced space hence we combined different dimension reduction methods with Slingshot as well. Palantir in contrast integrates dimension reduction step via diffusion maps to quantify the pseudotime progression from an early cell defined in advance. DDRTree, similarly, generates cell orderings by reducing the high-dimensional data to low-dimensional principle-tree structure, hence we have coupled DDRTree with preprocessing and normalization steps only. Furthermore, since Slingshot and DDRTree are unsupervised approaches, we have utilized Waddington-OT (WOT), a supervised approach that aims to find cell-cell transition probabilities at consecutive time-points via optimization of unbalanced transcriptional mass transfer. Comparison is somewhat imperfect however, as trajectories are defined slightly differently for each method. Since Slingshot estimates the smooth principal curve in low dimensional space, mapping of individual cells on the curve readily defines an ordering via the arc-length along the curve. In contrast, DDRTree embeds high-dimensional transcriptomic profiles onto a principal tree structure where the ordering is defined by the geodesic distances between individual cells. The supervised approach, WOT, on the other hand, generates a probability distribution between an individual cell at time ti and the cell population at time ti+1, hence the trajectory of an individual cell is defined as the vector of transition probabilities. In order to evaluate the results from different methods in a comparable fashion, we opted to use Spearman’s ρ which does not take into account the distances between individual cells, but rather only the orderings, hence different quantitative scales between methods can be compared.
Results
Dimension reduction and clustering
In order to evaluate how dimension reduction methods perform when coupled with the Leiden method for clustering we have compared the identified clusters using Adjusted Rand Index (ARI) across different subsets of gene and cell level thresholded datasets. However, note that since we do not have ground-truth observations of clusters, instead we have focused on the overlap of identified clusters between different methods to assess repeatability. Specifically, individual dimension reduction methods coupled with different preprocessing steps (imputation and normalization) are used to generate clustering via the Leiden method. Generated individual clusters are then compared using ARI and ARI values across different subsets are aggregated by taking the median of ARI values. This approach allowed us to investigate the stability of clusters for a given dimension reduction method when combined with different pre-processing steps. Furthermore, a common practice in scRNA-Seq analysis is preprocessing with Principal Component Analysis (PCA) to both reduce computational load and reduce variation/noise which requires selection of number of top latent features to keep where automated tools can be utilized. 52 However, dimension reduction via PCA can be non-trivial and introduce unwanted bias specifically in the case of multiple datasets hence we opted to not utilize PCA as an initial preprocessing step. As expected, we observed a positive correlation of ARI across different workflows with the number of cells (Supplemental Figure S1). However, ARI values showed reduced overlap between different methods across datasets globally, even when the number of cells is high (ARI < 0.75). Investigating methods individually showed t-SNE as relatively more robust to different preprocessing steps in the TKI dataset where remaining datasets showed variable performance (Supplemental Figure S2a-c). Interestingly, neural-network methods showed variable results where the use of DCA-NB/DCA-ZINB as a preprocessing step in the TKI dataset led to improved overlap between UMAP, UMAP + PAGA, and t-SNE. In contrast, the use of VAE as a dimension reduction method showed poor performance resulting in variable cluster assignments (See Supplemental Figure S3 eg, workflows). This suggests that as a dimension reduction method, neural-networks might not be the optimal choice but as a preprocessing step neural networks can provide advantages depending on the number of cells.
Datasets with relatively low number of cells showed variable results. For instance, in Pancreatic α cell differentiation, UMAP and PAGA + UMAP showed improved overlap when DrImpute is combined with Deconvolution but the overlap is reduced when ScTransform is used for normalization (Supplemental Figure S2f). The use of t-SNE similar to the TKI dataset was more robust to preprocessing steps. E2 Treatment dataset resulted in variable cluster assignments overall where both MCF7 and T47D cell line datasets resulted in different cluster assignments across workflows. The Neurodegeneration dataset on the other hand benefited from DCA with or without zero-inflation model but overall showed decreased overlap as well (Supplemental Figure S2h).
To further extend the analysis results, we have evaluated tooManyCells method as well which is another scRNA-Seq method used for clustering nearest-neighbor graphs to partition the cell population. 47 tooManyCells improved cluster overlap globally in the Pancreatic Maturation and Neurodegeneration datasets (Supplemental Figure S4). However, similar to Leiden clustering, selection of preprocessing workflows showed data-specific performance. For instance, the Pancreatic maturation α cells dataset was more sensitive to the imputation with DrImpute in contrasts with β cells dataset where imputation with DrImpute showed reduced overlap in cluster assignments when ScTransform is used for normalization (Supplemental Figure S4a). Interestingly, in the Neurodegeneration dataset, a dichotomy between the use of ScTransform and other workflows is observed where ScTransform showed poor overlap with other workflows (Supplemental Figure S4b).
We have also investigated the overlap of identified clusters with temporal information. Specifically, using homogeneity metric via R package clevr, we quantified the distribution of cells sampled from different time-points in a given cluster in order to delineate whether given methods can distinguish cells from different time-points. We observed a general improvement when TKI dataset is considered specifically when t-SNE, UMAP or PAGA-UMAP is applied where interestingly E2 treatment dataset showed the lowest homogeneity (Supplemental Figure S19). Furthermore, when ScTransform is coupled with DrImpute, substantial decrease in Pancreatic Maturation and Neurodegeneration datasets is observed which suggest that workflow selection should be done in a data specific fashion.
Trajectory estimation
In order to evaluate PTEs mapping to a latent biological process we used Spearman rank correlation and normalized entropy. As given previously, using rank correlation we aim to do a comparison of cell orderings identified by different workflows and normalized entropy is used to assess the distribution of rank correlations (bimodal around 0-1) in the case of Slingshot since >1 PTEs are identified (Supplemental Figure S5).
Slingshot
Evaluating the trajectories identified by Slingshot, we have observed large variation across different workflows and across different subsets. For instance, in Pancreatic maturation datasets, workflows that show relatively good overlap in α cell trajectories failed to identify overlapping trajectories in the β cell dataset. Specifically, the use of DrImpute or ScImpute resulted in decreased overlap of PTEs in β cell dataset (Figure 2). Furthermore, the number of cells did not correlate positively with the repeatability of identified trajectories where the majority of the workflows showed high entropy of rank correlations in the TKI treatment dataset with minimum entropy being >0.7 across 3 treatments (Supplemental Figure S6a-c). In contrast, datasets with relatively low number of cells showed slightly improved overlap for specific workflows. For instance, in the E2 treatment dataset, use of DM improved overlap in contrast with UMAP or UMAP + PAGA. The Neurodegeneration dataset on the other hand showed a global decrease in PTEs (Supplemental Figure S6).

Comparison of trajectories identified by Slingshot showing data dependent performance of each workflow. Combinations of icons for columns/rows represent distinct workflows. Entropy (upper triangle) is used to aggregate over multiple trajectories identified by Slingshot and data subsets corresponding to cell level and gene level filtering thresholds. Variation (lower triangle) over different data subsets is given to show the confidence for aggregating Entropy measure (See Supplementary Material for details). Results suggest data dependence where the use of imputation in β cells dataset significantly reduces the overlap of PTEs in contrast imputation step overall preserves the identified PTEs in α cells: (a) pancreatic differentiation α cells and (b) pancreatic differentiation β cells.
These results point out one of the major drawbacks of using Slingshot for PTEs; since the estimation of trajectories is heavily dependent on the prior dimension reduction step, heterogeneous datasets will necessarily show high variation to different parameter regimes. More specifically, the Slingshot method using principal curves can fail to capture the temporal dynamics on highly non-linear spaces hence need to be carefully selected/optimized for trajectory estimation. For instance, when UMAP is used for dimension reduction prior to PTE, the cell population structures remain overall similar as the number of cells increases but relative positioning of subpopulations can change in a way that does not reflect the latent temporal process (Supplemental Figure S7a-d). Furthermore, the non-linearity can artificially generate an increased number of trajectories resulting in diverge PTEs. For instance, use of DM resulted in 1 trajectory to be identified in E2 treatment data subsets hence resulting in “simpler” PTEs overall (Supplemental Figure S7g and h).
Palantir
We have also included an additional method widely used for pseudotime estimation. 49 Palantir utilizes nearest-neighbor graphs followed by diffusion maps as a dimension reduction/manifold learning step. Low dimensional representation is further used for pseudotime quantification as a distance measure from a defined progenitor cell. In order to marginalize out the selection of progenitor cell, we generate 10 pseudotime orderings using different progenitor cells sampled from initial time-point and calculate the average Spearman rank correlations. In the TKI dataset, Palantir showed relatively robust estimates of pseudotime orderings across different preprocessing steps where the average correlation remained >.5 (Supplemental Figure S8). However, similar to Slingshot results, data-specific overlap quality was present. For instance, the Alectinib treated dataset benefited from imputation by ScImpute across different subsets but Crizotinib and Lorlatinib treated datasets showed reduced overlap of PTEs. Furthermore, Crizotinib and Lorlatinib treated datasets showed distinct profiles for DCA-NB/DCA-ZINB where Crizotinib dataset benefited across different subsets from using DCA but Lorlatinib dataset showed subset dependent profile. In the Neurodegeneration and Pancreatic Maturation datasets, similar results were observed where ScTransform normalization helped improve the PTE overlap in the Neurodegeneration dataset but showed reduced overlap in the Pancreatic Maturation dataset specifically when coupled with DrImpute. Conversely, using DCA-NB/DCA-ZINB, Palantir PTEs showed relatively robust correlation across different workflows in the Pancreatic Maturation dataset (Supplemental Figures S9 and S10).
DDRTree
Since DDRTree/Monocle2 method inherently utilizes dimension reduction to generate a tree-like topology to define a latent trajectory, we have generated the combinatorial workflows for imputation and normalization steps only which is also reflected on the use of 2 icons instead of 3 where imputation is applied. However, also note that, in contrast with previous workflows, we have opted to further reduce the number of features by selecting top 50 principal components due to computational constraints hence the limitation of results to within method comparisons. We have aggregated rank correlations across 12 subsets by median values to evaluate the overlap of different workflows (Supplemental Figure S11). The TKI treatment dataset overall showed good overlap (ρ > 0.75) across different imputation and normalization methods. Interestingly however, Crizotinib treatment showed increased overlap of PTEs when DrImpute or ScImpute is utilized in comparison with when DCA is used (Figure 3a-c). Further investigating the individual trajectories showed that using DCA resulted in increased number of branch points in contrast with DrImpute or ScImpute (Supplemental Figure S12). This might be an implication for “overcorrection” when DrImpute or ScImpute is used subsequently reducing variation. Datasets with relatively low numbers of cells however showed variable results with different analysis steps having distinct “importance.” For instance, in the Neurodegeneration dataset, choice of normalization showed the highest impact where the use of Deconvolution decreased the trajectory overlap globally, in contrast, ScTransform was more robust to the imputation step (Figure 3d). Furthermore, as expected, E2 treatment dataset showed high correlation between workflows using Deconvolution and ScTransform normalization but not when DCA is used (Figure 3g and h) since, with low number of training dataset for the autoencoder model, parameters might not be estimated robustly. In contrast, pooling information from similar cells and genes might better capture biological signal. The Pancreatic differentiation dataset on the other hand showed increased overlap across different methods. Further investigating subset specific overlap of trajectories showed no substantial effect of gene or cell level quality filtering where the quality of overlap between different workflows remained similar across different subsets (Supplemental Figure S13).

Rank correlation of geodesic distances on DDRTree trajectories median aggregated over subsets showing both data specific performance and overall increase based on number of cells. (a-c) TKI treatment dataset shows improved overlap of cell orderings. Although the TKI dataset is relatively more heterogeneous, increased number of cells allow DDRTree to capture robust cell-cell similarities. (d-h) Remaining datasets show variable results with pancreatic maturation β performing comparable to TKI dataset and Neurodegeneration dataset performing the poorest.
WOT
Since both Slingshot and DDRTree aim to find a low dimensional ordering of individual cells in an unsupervised fashion, temporal information is not readily utilized which can lead to biased estimates where transcriptional dynamics are not “linearly” associated with time (Figure 4). Instead, supervised approaches can provide certain advantages for PTE by utilizing available temporal information. However, forcing individual cells in a supervised order also poses challenges such that cells are not synchronized in terms of division and growth rates. For this purpose, the WOT framework also allows us to calculate optimal growth rates for individual cells given the “transcriptional mass” transfer optimization problem. Furthermore, by removing the dimension reduction step, WOT inherently reduces the number of possible sources of variation. In order to evaluate how WOT performs when different methods for imputation and normalization are used, we have calculated pairwise rank correlations of transition probabilities between individual cells at consecutive time-points t0, t1, across different workflows. Simply, we have quantified how the transition probabilities of an individual cell change if a different normalization or imputation step is used.
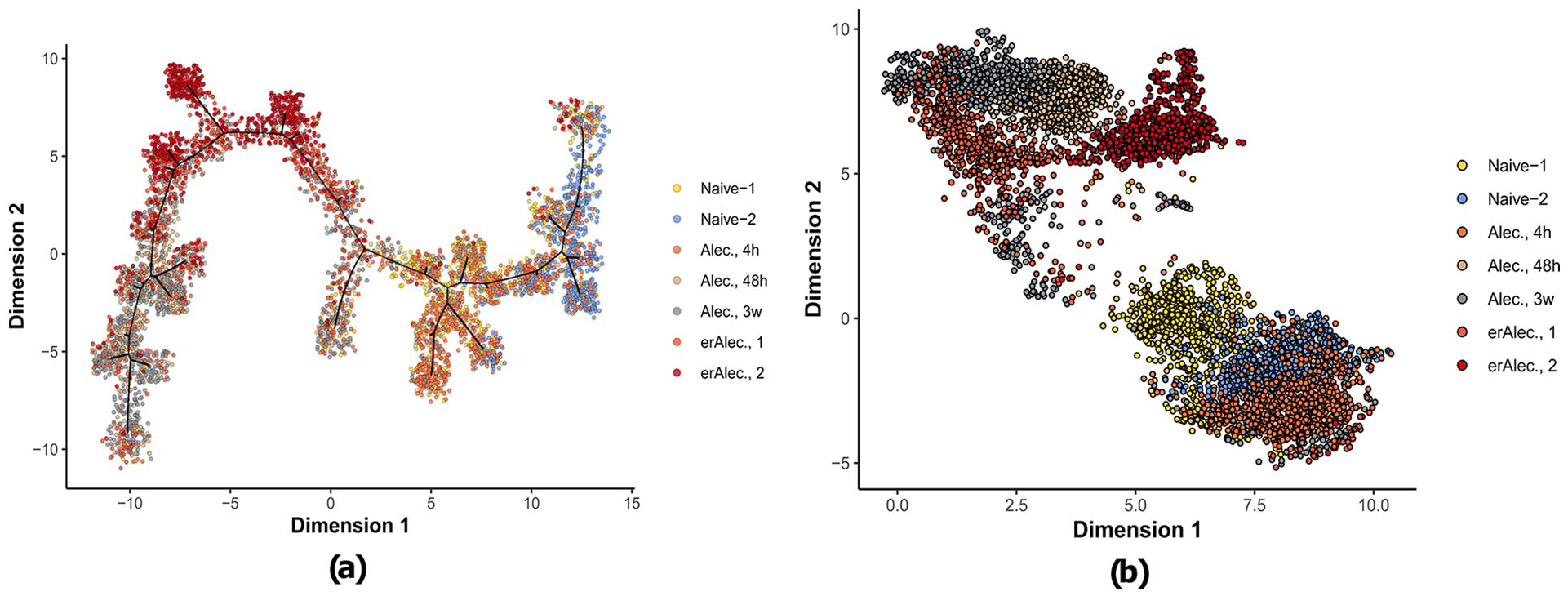
Sample dimension reductions for Alectinib treated NSCLC lines showing nonlinearity in temporal dynamics of gene expression. Since dimension reduction utilizes transcriptional similarity of individual cells, low dimensional representations might not necessarily correlate linearly with sampling time. In datasets where sampling time is not linear and/or the underlying transcriptional dynamics are highly heterogeneous supervised approaches might be more suitable where the change in transcriptional activity is ordered by the temporal process by default: (a) DDRTree and (b) PAGA-UMAP.
The TKI treatment dataset showed the highest overlap of transition probabilities across all pairwise workflow comparisons with median rank correlation >.75 (Supplemental Figure S14). Normalization with ScTransform showed slightly better overlap however when different imputation steps are compared in contrast with Deconvolution (Supplemental Figure S15). Interestingly however this difference was most striking in the Neurodegeneration dataset where the choice of imputation showed a relatively high difference of rank correlation (ρ > .2) between Deconvolution and ScTransform. Investigating imputation steps individually showed no substantial effect of imputation step where the overlap of trajectories when Deconvolution and ScTransform is used remained similar and relatively low (<.75) irrespective of which imputation step is used (Supplemental Figure S16). Investigating the effect of using different gene and cell level thresholds showed a substantial decrease in the Neurodegeneration dataset where the remaining datasets showed similar PTE comparison profiles across 12 subsets hence suggesting relatively robust PTEs across different threshold (Supplemental Figure S17). This suggests that WOT PTEs consistently show repeatable results specifically for datasets with relatively high number of cells captured (Supplemental Figure S18).
Discussion
With the advent of single-cell sequencing methods, identification of tumor subpopulations and pseudotime estimation has been extensively used where analysis of scRNA-Seq data is complicated by a multitude of factors. In order to evaluate methods developed for scRNA-Seq analysis we have aimed at evaluating the available methods in a combinatorial fashion to assess the repeatability of either identified subpopulations or estimated pseudotimes. We have shown that selection of different methods at different levels of scRNA-Seq analysis can lead to variable outcomes both for clustering and trajectory inference. This is especially important considering the availability of additional methods not utilized in this study and the continued proliferation of methods.53,54 Furthermore, we have observed substantial variation in workflows for either clustering or PTE where non-linear dimension reduction methods are used. This emphasizes the importance of careful evaluation of which methods to utilize since the results may not be generalizable to replicate datasets.
General trends in our analysis showed that the number of captured cells is crucial when deciding on which downstream analysis methods to use since datasets with relatively high number of cells can sample the evolutionary process on the underlying manifold more effectively hence showed increased overlap across different workflows specifically for clustering and PTE using WOT. Imputation approaches did not show improvement in downstream analysis as well which have been previously reported as well. 55 Dimension reduction methods that are heavily utilized in scRNA-Seq analysis showed high sensitivity to parameter selection hence clustering results using low dimensional representations were variable. Similar results were also shown previously. 56 Although, t-SNE and UMAP coupled with PAGA showed relatively robust cluster assignments there is no one best approach and methods showed data-specific performance. Clustering with tooManyCells on the other hand alleviated some of the limitations where clustering is done via nearest-neighbor graphs, however, data-specificity of workflows remained. This further stresses the importance of repeatability in scRNA-Seq analysis where unsupervised clustering is of major interest. In order to reduce some of the issues associated with clustering specifically when coupled with non-linear dimension reduction, “consensus” based approaches where randomly sampling features/cells might be more suitable.
Trajectory inference methods, similarly, showed variable results where non-linear dimension reduction is used. Slingshot for instance failed to capture reproducible trajectories in the TKI treatment dataset. As previously stated, Slingshot method based on principle curves is more suitable to relatively linear trajectories with a small number of branch points. Similar observations were also pointed out in previous studies as well. For instance, Saelens et al showed decreased performance of Slingshot when the underlying trajectory consisted of multiple branches and non-linearity, which as we have shown in this study, can be further exacerbated when considering multitudes of different preprocessing steps. In order to alleviate some of the issues in coupling dimension reduction methods with Slingshot, one may need to choose parameter regimes toward linearity, for instance increasing number of nearest neighbors or minimum distance parameter in UMAP. However, also note that the use of dataset from a single experimental setup is of limited applicability hence does not necessarily dismiss alternative views in the case of the TKI treatment dataset. Palantir on the other hand resulted in more robust PTEs across data subsets. This can be attributable to the fact that Palantir readily optimizes the number of dimensions to use to quantify PTEs hence reducing variation overall. Nevertheless, Palantir also suffered from data-specificity. Using either supervised approaches WOT or regularized dimension reduction using DDRTree resulted in increased correlations in trajectory estimates when different preprocessing methods are combined. DDRTree specifically showed improved performance over Slingshot especially when ScTransform is used for normalization but the quality of the overlap was data specific where TKI dataset with relatively large number of cells showed a global increase in correlation of identified trajectories. This is in contrast with Tian et al where Slingshot showed slightly improved performance over DDRTree. However, improved performance of Slingshot can be partially attributed to the mixture datasets being relatively less heterogeneous and the underlying structure being relatively linear.
Using supervised trajectory mapping via the WOT framework alleviated some of the issues with unsupervised approaches as well. Although identified trajectories remained sensitive to normalization method selection, data dependence is reduced where we have observed ScTransform performing relatively well across all the datasets. Furthermore, since the temporal information is utilized in WOT, we can readily assume the identified trajectories will overlap with the biological process compared to unsupervised alternatives. For instance, neither Slingshot nor DDRTree can differentiate subpopulations from different time-points if the transcriptional profiles are similar even though the temporal dynamics are different. However, it is also important to note that identified trajectories only regard the differences between individual cells in terms of transcriptional profiles mapped to low dimensional space (in the case of Slingshot and DDRTree). This makes the problem of evaluating the PTEs non-trivial due to absence of ground-truth observations Deviation from ground-truth PTEs should be evaluated using approaches that allow individual cells to be tracked.57,58 Furthermore, individual methods presented here can be further optimized separately resulting in improved PTEs. For instance, increasing the number of dimensions or using alternative metrics for quantifying transcriptional difference. Nevertheless, the WOT framework combined with ScTransform provided certain advantages by utilizing temporal information and reducing the variation.
In conclusion, analysis of scRNA-Seq datasets show high variation across different parameter regimes and methods in the context of clustering and trajectory mapping. It is non-trivial to utilize the heterogeneous structure of tumor subpopulations in order to extract biological insights hence analysis of scRNA-Seq requires careful selection of methods and optimization of parameters but different methods provide certain advantages. We hope that provided results can guide future studies for method selection and help with reproducibility in scRNA-Seq analysis.59-62
Supplemental Material
sj-pdf-1-evb-10.1177_11769343221123050 – Supplemental material for Stability of scRNA-Seq Analysis Workflows is Susceptible to Preprocessing and is Mitigated by Regularized or Supervised Approaches
Supplemental material, sj-pdf-1-evb-10.1177_11769343221123050 for Stability of scRNA-Seq Analysis Workflows is Susceptible to Preprocessing and is Mitigated by Regularized or Supervised Approaches by Arda Durmaz and Jacob G Scott in Evolutionary Bioinformatics
Footnotes
Acknowledgements
This work made use of the High-Performance Computing Resource in the Core Facility for Advanced Research Computing at Case Western Reserve University.
Funding:
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Institutes of Health (5R37CA244613-02) and the American Cancer Society Research Scholar Grant (RSG-20-096-01).
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
AD—Conceptualization, Implementation, Analysis, Manuscript Preparation. JGS—Review and Editing, Project Supervision. All authors have read and approved the final manuscript.
Availability of Data and Materials
All the datasets utilized in the study are publicly available from corresponding resources.
TKI Treatment Dataset is not openly available due to the decisions of original study authors but is available upon request from original publication. 31
Mouse model of pancreatic islet αβ cell maturation dataset is openly available from Gene Expression Omnibus with accession id GSE87375 and repository link for raw data (![]() ).
).
Neurodegeneration Dataset is openly available from Gene Expression Omnibus with accession id GSE103334 and raw data is provided as supplementary from the original publication.
ER+ Breast Cancer E2 Treatment Dataset is publicly available from Gene Expression Omnibus with accession ids GSE107858 and GSE107863 for MCF7 and T47D respectively with repository links for raw data (https://www.ncbi.nlm.nih.gov/Traces/study/?acc=PRJNA418865o=acc_s%3Aa) and (![]() ).
).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.