
Abstract
Hepatitis B virus (HBV) infection is a major cause of acute liver failure (ALF) in China, and mortality rates are high among patients who do not receive a matched liver transplant. This study aimed to determine potential mechanisms involved in HBV-ALF pathogenesis. Gene expression profiles under access numbers GSE38941 and GSE14668 were downloaded from the Gene Expression Omnibus database, including cohorts of HBV-ALF liver tissue and normal samples. Differentially expressed genes (DEGs) with false discovery rates (FDR) <0.05 and |log2(fold change)| >1 as thresholds were screened using the Limma package. Gene modules associated with stable disease were mined using weighed gene co-expression network analysis (WGCNA). A co-expression network was constructed and DEGs were analyzed using gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses. A gene-based network was constructed to explore major factors associated with disease progression. We identified 2238 overlapping DEGs as crucial gene cohorts in ALF development. Based on a WGCNA algorithm, 10 modules (modules 1-10) were obtained that ranged from 75 to 1078 genes per module. Cyclin-dependent kinase 1 (CDK1), cyclin B1 (CCNB1), and cell-division cycle protein 20 (CDC20) hub genes were screened using the co-expression network. Furthermore, 17 GO terms and 6 KEGG pathways were identified, such as cell division, immune response process, and antigen processing and presentation. Two overlapping signaling pathways that are crucial factors in HBV-ALF were screened using the Comprehensive Toxicogenomics Database (CTD). Several candidate genes including HLA-E, B2M, HLA-DPA1, and SYK were associated with HBV-ALF progression. Natural killer cell-mediated cytotoxicity and antigen presentation contributed to the progression of HBV-ALF. The HLA-E, B2M, HLA-DPA1, and SYK genes play critical roles in the pathogenesis and development of HBV-ALF.
Keywords
Background
Acute liver failure (ALF) is a rapid loss of liver function that comprises massive hepatocellular necrosis leading to multiorgan failure. Patients with ALF develop hepatic encephalopathy and impaired protein synthesis within a few weeks. Over the past few decades, ~2000 patients per year have experienced ALF in the United States. 1 The etiology of ALF varies between different geographic regions. Infection with hepatitis A, B, and E viruses is the most prevalent cause of ALF in developing countries, whereas drugs are the main causes in the United States and western Europe.2,3 Hepatitis B virus (HBV) is a major cause of ALF in China, and overall mortality has historically exceeded 80%.1,4 Liver transplantation and intensive clinical care have significantly improved survival in recent years; 2-year survival rates have increased to >90% for patients after liver transplantation. 5 Although considerable progress has resulted in a better understanding of ALF, the pathogenesis of HBV-ALF remains unclear. Thus, the early detection, accurate diagnosis, and prognostic evaluation of ALF should be improved.
Advanced gene sequencing has led to the discovery of novel mechanisms that are involved in disease progression. Immune injury, ischemic hypoxia, and endotoxin-induced damage are currently recognized as major factors in the process of liver injury associated with ALF. 6 Gene profiling and histological findings 7 have shown that that humoral immunity against HBV core antigen might play key roles in the pathogenesis of ALF. Gene profiling has also shown that abnormal gene expression in HBV-ALF samples correlates with hepatic stem/progenitor cell activation and fibrogenesis. 8 Bioinformatic analysis of the microarray data sets GSE38941, GSE62029, GSE62030, and GSE14668) by Lin et al 9 associated the genes ORM1, ORM2, PLG, and AOX1 with the immune response, as well as the complement and coagulation cascade pathways that might participate in the pathogenesis of HBV-ALF. However, studies of gene expression associated with HBV-ALF have remained relatively scant.
We integrated previous microarray data to determine the potential mechanism(s) of HBV-ALF. Larger cohorts of differentially expressed genes (DEGs) were identified in HBV-ALF than in normal liver samples. We mined gene modules using weighed gene co-expression network analysis (WGCNA), constructed a co-expression network, and analyzed functional enrichment. We identified two signaling pathways (natural killer [NK] cell-mediated cytotoxicity and antigen processing and presentation) and the HLA-E, B2M, HLA-DPA1 genes as factors that might be involved in HBV-ALF progression. Our findings suggested that immune-biological processes (BPs) play major roles in the pathogenesis and development of HBV-ALF.
Materials and Methods
Data resources
Information was retrieved on May 24, 2019. We searched all publicly uploaded genomic profiles in the NCBI GEO 10 (http://www.ncbi.nlm.nih.gov/geo/) database using the keywords “hepatitis B virus,” “acute liver failure,” and “Homo sapiens.” The genomic expression profiles should include HBV-ALF and normal liver samples, and number of specimens should be more than 20. Finally, we obtained 2 data sets under the access numbers GSE389418 and GSE14668. 1 The GSE38941 data set included 17 liver tissue samples derived from patients with HBV-ALF and 10 normal liver samples. The GSE14668 data sets consisted of 8 HBV-ALF liver specimens and 12 normal liver specimens from healthy donors. These data sets were tested using the platform of the [hg-u133_plus_2] Affymetrix Human Genome U133 Plus 2.0 Array.
Screening DEGs associated with HBV-ALF
The original format of the two microarray data sets was downloaded from the Affymetrix platform. The data set was normalized using the Oligo package 11 in R3.4.1 version 3.6 (http://www.bioconductor.org/packages/release/bioc/html/oligo.html) followed by background correction (using the MAS method), format conversion, missing value complement, and data normalization. We then screened and compared DEGs associated with HBV-ALF and control samples using the Limma package 12 version 3.34 (https://bioconductor.org/packages/release/bioc/html/limma.html). A false discovery rate (FDR) <0.05 and |log2 fold change (FC)| >1 were the selected thresholds. Hierarchical clustering was assessed using the Pheatmap package 13 version 1.0.8 (https://cran.r-project.org/web/packages/pheatmap/index.html) based on Euclidean distance methods 14 and the results were visualized in a heatmap.15,16 We identified DEGs that overlapped between the GSE38941 and GSE14668 data sets. For these common genes, Gene ontology (GO) BPs and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment were analyzed using the online Database for Annotation, Visualization and Integrated Discovery (DAVID) version 6.8 (https://david.ncifcrf.gov/).17,18
Screening stable gene modules using WGCNA algorithm
The bioinformatics method WGCNA has been extensively used to construct gene co-expression networks and mine gene modules associated with stable diseases. 19 We used the WGCNA package 20 version 1.61 in R3.4.1 (https://cran.r-project.org/web/packages/WGCNA/index.html) to mine HBV-ALF-related gene modules. The data sets GSE38941 with a relatively large number of samples served as the training set, and the GSE14668 data set served as the verification set. Gene expression status in the two data sets was compared and correlations among values were analyzed. Meanwhile, the adjacency function was defined, and gene module divisions and stability were evaluated. Modules with >80 genes were obtained and the cutHeight was set at 0.99.
Co-expression network construction
Interactions between candidate DEGs and the constructed co-expression network were explored using STRING 21 version 10.0 (http://string-db.org/). The network was visualized using Cytoscape version 3.6.122 (http://www.cytoscape.org/). We screened crucial DEGs associated with HBV-ALF by calculating the topology parameters of nodes (degree, betweenness centrality, and closeness centrality). The functional enrichment of these DEGs was analyzed.
Candidate target gene screening for HBV-ALF
We used the keywords “acute liver failure” to search the Comprehensive Toxicogenomics Database 23 (2019 update, http://ctd.mdibl.org/) for genes in the KEGG pathway that are closely associated with HBV-ALF. After comparisons with previous pathways, we screened overlapping signaling pathways as key factors in disease progression as well as associated DEGs.
Results
Screening DEGs associated with ALF
The information derived from the two data sets was normalized. After setting the threshold in the GSE38941 data sets, we screened 2558 DEGs from HBV-ALF samples and compared them with normal liver tissue samples, including 685 and 1873 that were, respectively, downregulated and upregulated. We screened 5317 DEGs from the GSE14668 data set that including 1042 and 4275 that were downregulated and upregulated, respectively. The distribution of DEGs was visualized using volcano plots, and Figure 1 shows a bidirectional hierarchical clustering heat map.
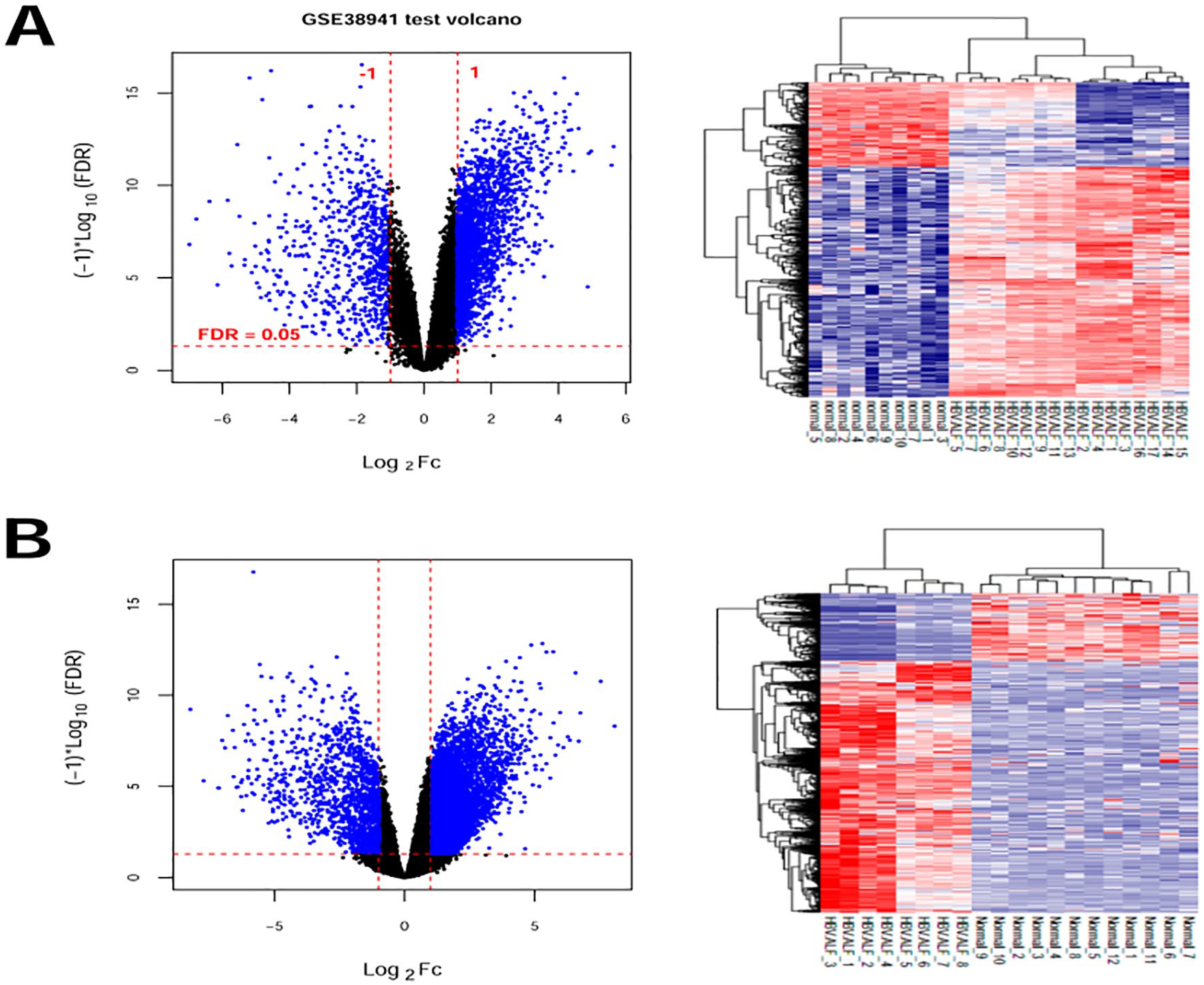
Volcano plots and supervised hierarchical cluster heat map of DEGs between hepatitis B virus-associated acute liver failure and control liver samples. Blue dots, DEGs in GSE38941 (A) and GSE14668 (B) data sets. Red horizontal lines, FDR < 0.05; vertical lines, |log2FC| > 1. DEGs, differentially expressed genes. FDR, false discovery rate.
We screened 2238 overlapping genes as crucial gene cohorts associated with ALF disease (Figure 2A). Most genes represented consistent expression status (2236 consistent DEGs), whereas the expression of only 2 genes contrasted. Functional enrichment of these 2236 overlapping DEGs was analyzed. We screened 84 GO-BP terms and 50 pathway categories. Figure 2 shows the top 20 terms according to the FDR. These DEGs were mainly enriched in GO-BP terms associated with regulation of the immune response, the inflammatory response and neutrophil-mediated immunity. The major pathway categories included cell adhesion molecules, complement, and coagulation cascades.

Crucial DEGs screening and function enrichment. (A) Venn diagram based on overlapping DEGs between GSE38941 and GSE14668 data sets. (B) GO terms and, (C) KEGG pathway analysis for DEGs associated with acute liver failure. Horizontal axis, number of genes; vertical axis, GO terms or pathway categories. Orange columns indicate higher significance. DEGs, differentially expressed genes; GO, gene ontogeny; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Mining gene modules according to WGCNA algorithm
Here, we used the WGCNA algorithm to further mine the functional gene modules associated with ALF disease. The results showed that correlations of both expression status and connectivity were positive between the 2 data sets (cor = 0.9; P < 1e–200; cor = 0.57; P = 7.5e–193).
We explored the value of the soft-thresholding power that represented the adjacency matrix weight parameter to fulfill the scale-free network topology (R 2 > 0.9). Subsequently, interaction coefficients (coef) between log (k) and logp (k) were calculated. Higher coef values indicated that the co-expressed network had closer scale independence (Figure 3B). The power value was set to 36, then mean connectivity as a function of soft-thresholding power was evaluated (Figure 3B).

Co-expression analysis of DEGs in two data sets and definition of adjacency function: (A) scatter diagram shows ranked gene expression (left) and connectivity (right) in two data sets. (B) Definition of weighted parameter power in adjacency function. Left panel, scale-free topology model; right panel, mean connectivity as function of soft threshold. Red line in left panel represents the squared value of a correlation coefficient of 0.9.
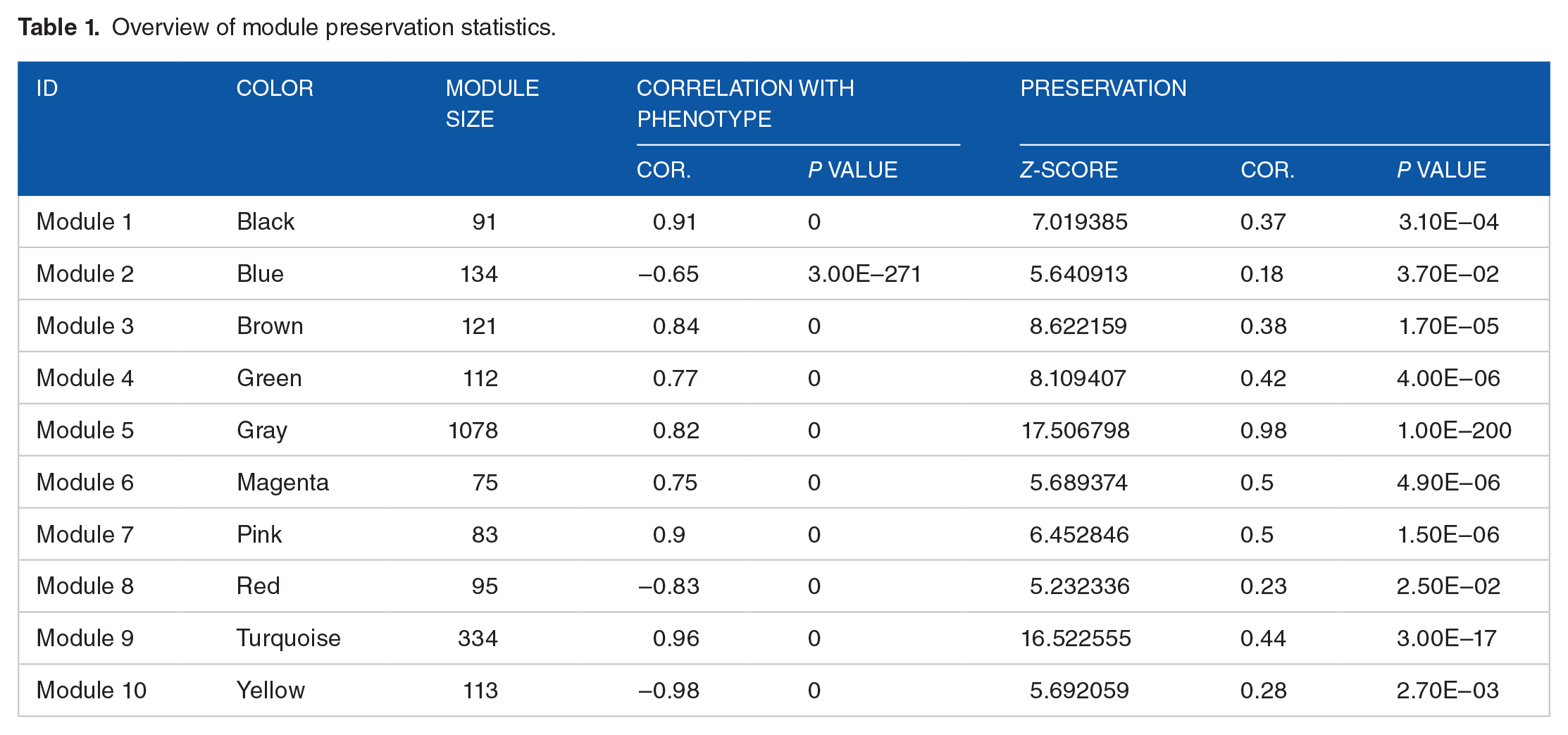
To further screen the gene modules, we used cluster dendrograms to detect co-expressed clusters based on specific thresholds (cutHeight = 0.99; number of genes ⩾ 80). Figure 4A shows the clustering results of mined modules in the GSE38941 (upper) and GSE14668 (lower) data set. We obtained 10 modules (module 1-10) comprising 75 to 1078 genes. Entire modules were assigned specific colors (yellow, blue, red, brown, green, black, turquoise, magenta, pink, and gray). Table 1 shows a complete list of preserved modules, which was high for whole modules (Z-score > 5; P ⩽ 0.05). The 1078 genes that were not assigned to functional modules are colored gray. The heatmap in Figure 4B shows relationships between gene modules and disease traits. The coefficient values for the black, brown, pink, and turquoise modules were the same as those of control gray modules. Thus, a total of 629 DEGs in the 4 modules were identified as candidate genes associated with the development of HBV-ALF (Figures 5 and 6).

WGCNA of hepatitis B virus-associated acute liver failure-related genes: (A) clusters of mined modules in GSE38941 (upper) and GSE14668 (lower) data sets. (B) Module-trait relationships. Colors ranging from blue to red represent change from negative, to positive correlation. WGCNA, weighed gene co-expression network analysis.
Overview of module preservation statistics.

Construction of co-expression network and functional enrichment analysis: (A) construction of protein-protein interaction network. Color on node edge corresponds to module with same color and node size is shown in degrees. (B) Gene ontogeny terms and KEGG signaling pathway enrichment analysis of DEGs in co-expression network. Colored dots with different sizes indicate significance of enrichment results. DEGs, differentially expressed genes; KEGG, Kyoto Encyclopedia of Genes and Genomes.
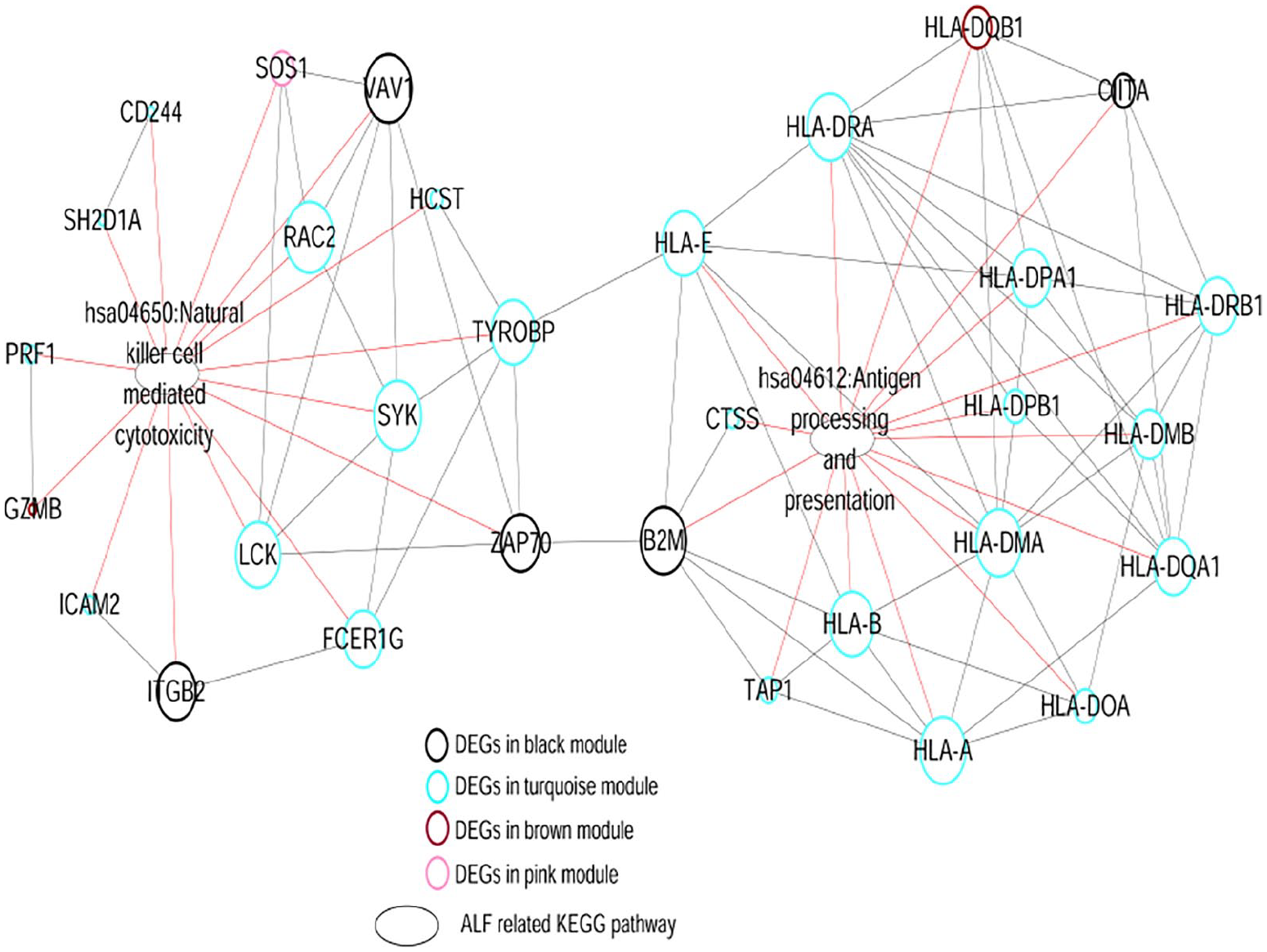
Comprehensive network construction of crucial signaling pathways and DEGs associated with HBV-ALF. Color on node edge corresponds to module with same color. Oval node, KEGG signaling pathway. Black and red lines, interactions between genes and pathways.
Construction of co-expression network
A co-expression network was constructed for these 629 DEGs according to the STRING database. Interaction pairs with scores >0.8 were reserved and a co-expression network comprising 849 edges and 214 gene nodes was constructed. All of these DEGs were notably upregulated in ALF specimens. Hub genes were identified by evaluating the topological parameters of node degree, betweenness centrality, and closeness centrality. The top 10 hub genes listed in Table 2 include cyclin-dependent kinase 1 (CDK1; degree = 62), cyclin B1 (CCNB1; degree = 48), cell-division cycle protein 20 (CDC20; degree = 47). Moreover, analysis of the functional enrichment of these DEGs using the online tool DAVID identified 17 GO-BP terms and 6 pathway categories (Table 3). These DEGs were mainly involved in cell division, immune response process, antigen processing and presentation, and NK cell-mediated cytotoxicity signaling pathways.
Topological parameters of top 10 hub genes in protein-protein interaction network.

GO terms and KEGG pathways enrichment analysis for differential expressed genes associated with acute liver failure.

Screening candidate target genes in CTD database
We searched the CTD database using the keyword “acute liver failure” to explore potential mechanisms of HBV-ALF and found 110 KEGG signaling pathways. Two overlapping pathways, namely antigen processing and presentation, and NK cell-mediated cytotoxicity were identified after comparisons with the previously determined pathways. A gene-pathway interaction network was constructed that consisted of two pathways and several DEGs, including HLA-E, HLA-DPA1, TYROBP, SYK, LCK, RAC2, and ZAP70. The results indicated that these DEGs and associated pathways are crucially involved in the development of HBV-ALF.
Discussion
We investigated changes in genes associated with HBV-ALF processes using the WGCNA algorithm. Genomic profiling identified numerous DEGs between HBV-ALF and normal liver tissue samples. Four gene modules were selected for detailed analysis and 629 related DEGs were identified as being vitally involved in ALF processes. We constructed a co-expression network and considered CDK1, CCNB1, and CDC20 as hub genes in disease development. We also found that the signaling pathways, NK cell-mediated cytotoxicity, and antigen processing and presentation, were involved in HBV-ALF. Several genes, namely, HLA-E, B2M, HLA-DPA1, TYROBP, ZAP70, and SYK might be candidates in HBV-ALF progression, because abundant NK cells in the human liver comprise the major effector population in the innate immune system. Activation of the NK-cell-mediated cytotoxicity pathway during HBV infection could result in liver damage and contribute to the development of HBV-ALF. 24 However, the potential mechanism has remained obscure. Others have shown that during the processes of HBV-ALF or FHF, the KCTD9, Fas/FasL, NKG2D/NKG2D ligand and TRAIL pathways25-27 interact with the NK cell-mediated cytotoxicity pathway and contribute to hepatocyte injury. This study identified 16 DEGs that were associated with NK-cell-mediated cytotoxicity pathways during disease progression. Among these, RAC2, SYK, TYROBP, ZAP70 had a highly connective degree in the comprehensive network. RAC2 encodes a GTP-metabolizing protein that belongs to the Rac family and the activity of Rac in leukocytes is essential for immunity. Depletion of RAC2 (but not RAC1) can induce mitochondrial dysfunction in human leukemic stem/progenitor cells. 28 Patients with mutations of the RAC2 gene have the clinical features of a common variable immunodeficiency. 29 A recent study showed that RAC1/RAC2 and SFK can regulate the NK cell-mediated cytotoxicity process through PI3K activation for defense against bacterial infection. 30 TYROBP, also known as DAP12, encodes a transmembrane signaling polypeptide. The signal adapter, DNAX-activating protein of 12 kDa (DAP12) was initially described as an immunoreceptor containing a tyrosine-based activation motif and phosphorylated DAP12 peptides interact with zeta-chain-associated protein kinase (ZAP)-70 and spleen tyrosine kinase (SYK) that are involved in activating NK cells. 31 An enzyme belonging to a tyrosine kinase family encoded by ZAP70 regulates T-cell development and lymphocyte activation. Spleen tyrosine kinase is involved in the immune cell activation process, and HBV or HCV infection can significantly increase SYK and related cytokine expression in hepatocytes, thus resulting in the development of liver fibrosis. 32 Pugh et al 33 showed that human NK cells can downregulate Syk and Zap70 kinases in response to prolonged activation or DNA damage. Regulating NK cell activity has proven valuable as an immunotherapeutic strategy against various disease states.
Moreover, the antigen processing and presentation pathway was abnormally expressed during disease progression and 15 related DEGs were enriched in this pathway. Antigen processing is an immunological process that prepares antigens for presentation to specialized immune cells, which is essential for the T-cell immune response. The potential mechanism of antigen processing and presentation machinery in HBV-ALF is still not clear. We selected several associated candidate genes for detailed analysis. Human HLA-E is a nonclassical MHC class I molecule, and less of it is expressed on cell surfaces compared with classical paralogues. Infection with HCV results in chronic inflammatory changes in liver tissues that lead to fibrosis and cirrhosis. The expression of HLA-E is significantly upregulated in patients infected with HCV at the severe stages of liver fibrosis. 34 Genome-wide sequencing has shown that B2M is mutated in several types of cancers, whereas little is known about its status in HBV-ALF.35-37 The B2M gene encodes beta-2-microglobulin protein (B2M), which is a human leukocyte antigen (HLA) domain. Class I HLA antigens play major roles during the process of hepatitis virus infection by eliminating the virus. Serum levels of B2M are high in patients with chronic active HBV and in those with liver cirrhosis associated with HCV.38,39 We found that abnormally expressed B2M is involved in antigen processing and presentation pathways, indicating that B2M might be a marker of disease progression in HBV-ALF.
A recent study has applied similar approaches to analyze the GSE38941, GSE62029, GSE62030, and GSE14668 microarray data sets. 9 That study showed that the ORM1, ORM2, PLG, and AOX1 genes that are associated with the immune response and complement and coagulation cascade pathways might participate in the pathogenesis of HBV-ALF. Although the approaches were similar, the thresholds for screening DEGs differed. The criteria of|fold change| ⩾ 2 and FDR < 0.01 were applied in that study, which generated 624 and 1395 DEGs for GSE38941 and GSE14668, respectively. However, we selected DEGs based on the thresholds of |log2 fold change (FC)| > 1 and FDR < 0.05, and identified 2258 and 5317 DEGs in GSE38941 and GSE14668, respectively. We used relatively looser criteria to collect more DEGs for WGCNA analysis. In addition, although the hub genes in this study and in that of Lin et al 9 were identified from protein-protein interaction networks, the methods used to build the networks differed. The 629 DEGs that we used to build the network were in the black, brown, pink, and turquoise modules, and had higher coefficients with disease traits. These genes might be associated with HBV-ALF development.
Although this study provided a comprehensive network of pathways and crucial genes, some limitations persisted. First, we did not have a large number of clinical liver samples. Second, the potential roles of candidate genes in the development of HBV-ALF disease should be experimentally validated.
Conclusion
We identified several crucial DEGs associated with HBV-ALF, such as CDK1, CCNB1, HLA-E, B2M based on the WGCNA method and a PPI network. Our findings suggested that DEGs involved in two signaling pathways are key factors in disease progression. The present findings provided novel insight and potential therapeutic targets for patients with HBV-ALF.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
DCQ designed the study. YS and HTY collected the data and were major contributors in drafting the manuscript. FFL, LQL, DXH, and HJZ analyzed and interpreted the data. DCQ revised the manuscript for critical content. All authors read and approved the final version of the manuscript.