
Abstract
In polymerase chain reaction (PCR)-based DNA sequencing studies, there is the possibility that mutations at the binding sites of primers result in no primer binding and therefore no amplification. In this article, we call such mutations PCR dropouts and present a coalescent-based theory of the distribution of segregating PCR dropout mutations within a species. We show that dropout mutations typically occur along branch sections that are at or near the base of a coalescent tree, if at all. Given that a dropout mutation occurs along a branch section near the base of a tree, there is a good chance that it causes the alleles of a large fraction of a species to go unamplified, which distorts the tree shape. Expected coalescence times and distributions of pairwise sequence differences in the presence of PCR dropout mutations are derived under the assumptions of both neutrality and background selection. These expectations differ from when PCR dropout mutations are absent and may form the basis of inferential approaches to detect the presence of dropout mutations, as well as the development of unbiased estimators of statistics associated with population-level genetic variation.
Introduction
DNA sequencing is either primer based, whereby a specific genetic locus is amplified by polymerase chain reaction (PCR) and sequenced 1 or by shotgun sequencing, whereby genomes are randomly broken into small segments and sequenced directly with no amplification.2,3 In primer-based sequencing, 2 primers that together flank a locus are used. Primers are typically about 18 to 24 nucleotides in length and bind to DNA. Each primer initiates replication on separate strands, with complementary effects such that after several rounds of replication and strand separation a specific locus is amplified. 4
Substitutions at the binding sites of primers can lower the probability a primer binds and initiates replication. Not all substitutions at the binding site of a primer are equal. For example, substitutions within the last 3 to 4 nucleotides of the 3′ end can significantly reduce PCR replication.5,6 Primer coverage corresponds to the proportion of species or taxa in the sample that are, or are expected to be, amplified and sequenced for a primer or set of primers. 7 Studies of primer coverage that compare primers to DNA sequence databases indicate that coverage for a specific primer varies depending on taxon, ranging from 0% to nearly 100%,7,8 where the taxon level is at the domain or phylum. For example, in the bacterial phylum, Nitrospirae coverage is about 97% for the commonly used 16S rRNA primer 519F if a single mismatch is allowed within the last 4 nucleotides of a primer binding site, but only about 32% if a single mismatch is not allowed. 7 For the bacterial phylum Lentisphaerae and the commonly used 16S rRNA primer 338F, coverage is 97% if a single mismatch is allowed within the last 4 nucleotides of a primer binding site, but 0% if a single mismatch is not allowed. 7 High coverage may occur at the phylum level for a specific primer, but is typically lower at the domain level due to broader sequence diversity.
A lack of primer coverage is straightforward to detect in studies where it is sought to sequence DNA from a single individual because amplification and sequencing will fail, which is directly detectable. In contrast, it is becoming increasingly common to pool, amplify, and sequence DNA from multiple individuals across one or more species. For example, in primer-based metagenomic and eDNA studies, a sample is taken from the environment that contains individuals and/or DNA from potentially thousands of species or OTUs (operational taxonomic units) and multiple individuals per species or OTU. In this approach, PCR amplification and sequencing is often non-targeted and used to discover or detect the species, OTUs or higher taxonomic groups present, as well as targeted and used to detect a specific species, OTU or taxonomic group. In non-targeted applications, there is the possibility that a taxonomic group or subset of individuals within a taxonomic group go undiscovered or undetected because of a mismatch between primers and binding sites. Furthermore, even with targeted sequencing, there is the possibility of low primer coverage.7,8
As context, many datasets deposited in the European Bioinformatics Institute metagenomics database (https://www.ebi.ac.uk/metagenomics/) 9 involve primer/amplicon-based and non-targeted sequencing from nature. Although there has been progress in developing reference databases for primer/amplicon-based metagenomic studies to match sequences to species or higher taxonomic groups in a targeted manner, 10 there is still a need for assessing whether a taxonomic group goes partially or fully unsequenced in non-targeted sequencing studies, as well as assess the degree of coverage of targeted taxa.
A challenge for primer-based metagenomic studies is assessing coverage for a sample from nature. A rarefaction curve may be used to assess coverage by plotting the number of taxa delineated versus the number of sequences recorded. 11 If this curve is asymptotic, then nearly all taxa have been delineated. Nevertheless, this only applies to taxa that are amplified by the primer or primer set. Even if primer coverage was less than 100%, an asymptotic rarefaction curve can occur with more sequences because eventually all amplified species can eventually be sequenced and delineated to a taxon or as coming from an unknown taxon. Generally, it would be helpful for a metagenomic study to have available approaches to assess primer coverage for their specific sample from nature. In this article, we present theory to help the development of approaches to assess coverage.
In particular, we use coalescent theory to derive expectations for the pattern of nucleotide variation within a species when a mutation or a set of mutations at primer site(s) block the chain of events during a primer-based sequencing study, such that a set of sequences are not recorded as being present. This mutation or a set of mutations could either completely block a primer from binding to its binding site or reduce binding, such that amplification is so low that the locus is not sequenced for a set of individuals descendent from the mutation or a set of mutations. We call such mutations “PCR dropouts.”
A PCR dropout mutation or a set of PCR dropout mutations may occur ancestrally to all individuals within a species or later (forward in time), such that only a subset of individuals from a species have one or more of the dropouts. Dropout mutations that occur ancestrally to all individuals and are sufficient to block amplification of all individuals within the species are not the focus of this study. Instead, we focus on dropouts that are segregating within a population, such that these dropouts occur later (forward in time) than the most recent common ancestor (MRCA) to the species. Using coalescent theory,12,13 we show that segregating dropout mutations give rise to distinct patterns of DNA sequence variation. This pattern may be used to assess whether a species (or OTU) is prone to PCR dropouts and therefore reduced coverage. Furthermore, if several species (or OTUs) within a taxon have a signal of dropouts, this may indicate a larger problem of coverage for the entire taxon.
Although this article focuses on dropouts within a species, similar principles apply at higher levels of biological organization. We choose to first focus on within-species variation because DNA sequence evolution is well characterized by the coalescent process and this process is universal among species and taxa. In contrast, at higher levels, there is no standard theoretical framework of DNA sequence evolution, although Hey’s 14 approach may be promising in the context PCR dropouts and metagenomics (see section “Discussion”).
Expectations for DNA sequence variation at the population level and in the presence of segregating dropout mutations will also be of potential use in other contexts besides assessing primer coverage. For example, there has been the development of estimators of nucleotide diversity that account for the higher rate of sequencing errors associated with next-generation sequencing technology.15,16 Yet, methods are not available that account for segregating PCR dropouts. Furthermore, methods have been developed to use population-level variation to infer the phylogenetic structure of ecological communities using metagenomic data. 17 Accordingly, it would be helpful to consider the effect of PCR dropouts on population-level variation and whether they may affect inferences of phylogenetic structure.
The organization of this article is as follows: First, we present theory assessing the probability that dropout mutations occur along branch sections that are deep in the coalescent tree of a species or along branch section that occur more recently, as well as whether we expect a species to segregate a single dropout mutation or multiple dropout mutations. Generally, we find that dropout mutations typically occur along branch sections that are deep in the coalescent tree of a species. Furthermore, given rates of mutation at primer sites, a single dropout mutation typically segregates in a sample. Focusing on dropouts that completely block amplification, we then derive expectations for the distribution of coalescence times and pairwise differences of sequenced alleles at a locus. This analysis indicates that the distribution of coalescence times and pairwise differences is affected by the presence of a dropout and may therefore give rise to a detectable signal of a dropout. The theory accounts for the process of background selection,18,19 which is occurring at loci used in primer-based metagenomic studies, such as 16S/18S RNA. 1
Theory
It is assumed that a sample is taken from an environment, be it water, soil, feces, etc, and DNA is purified from the sample and subject to PCR and sequencing. Although it may be that within a sample there are individuals across the tree of life, only a single genetic locus is amplified by PCR. For simplicity, we assume that the PCR primers target bacteria and archaea such that species are haploid and effective population sizes are potentially large. Although this article focuses on haploid microbes, the theory applies directly to diploid microbes with a change in timescale (see below). For a given species in the sample, there is a sample size of n. This sample size is not known. In the theory presented, we nevertheless assume a sample size of n and then develop expectations for how many individuals within the sample are expected to be unamplified given a sample of size n. The theory demonstrates that n is a nuisance parameter and detectable effects of dropout mutations can be inferred without the knowledge of n.
For PCR sequencing of a species or OTU to occur, DNA primers need to amplify the DNA of a species or OTU to a sufficient level that allows for sequencing. Here we define
Neutrality and no background selection at the amplified locus
A natural theoretical framework to model dropouts assuming neutrality is Kingman’s
12
coalescent (see also Tajima
13
). In the Kingman coalescent, the genealogical history of a sample of size n is divided into
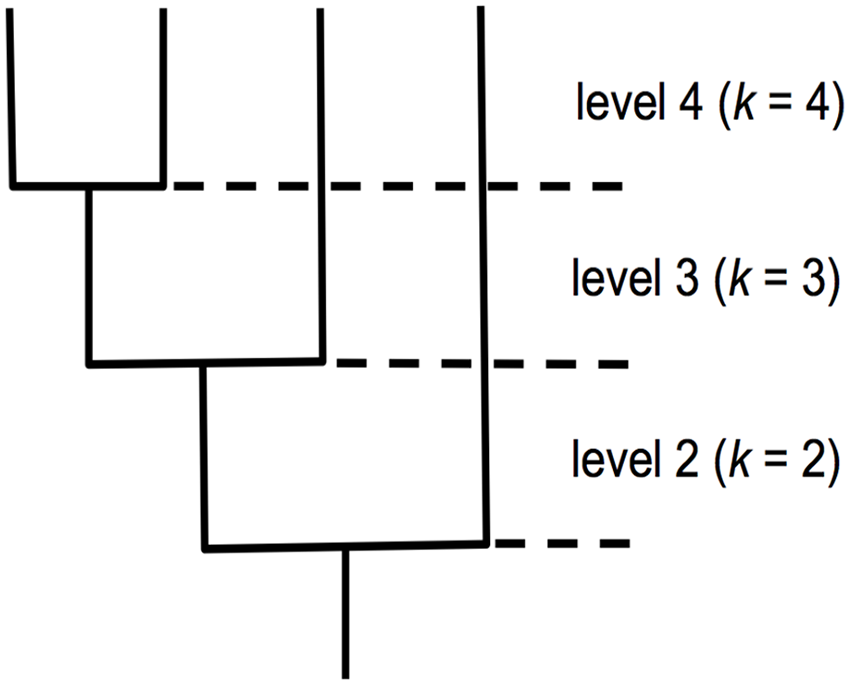
The use of the term “level.” A level corresponds to the number of branch sections in the coalescent tree. Branch sections are not drawn to scale. Lower levels in the tree have longer branch sections according to coalescent theory. 12

The use of the term “unique.” In the coalescent tree, 2 branch sections at level 4 have a dropout mutation, such that
Probability of
branches with dropouts at level k,
Define

where to simplify notation we let
The expected number of dropout mutations per branch section at level k is

noting that
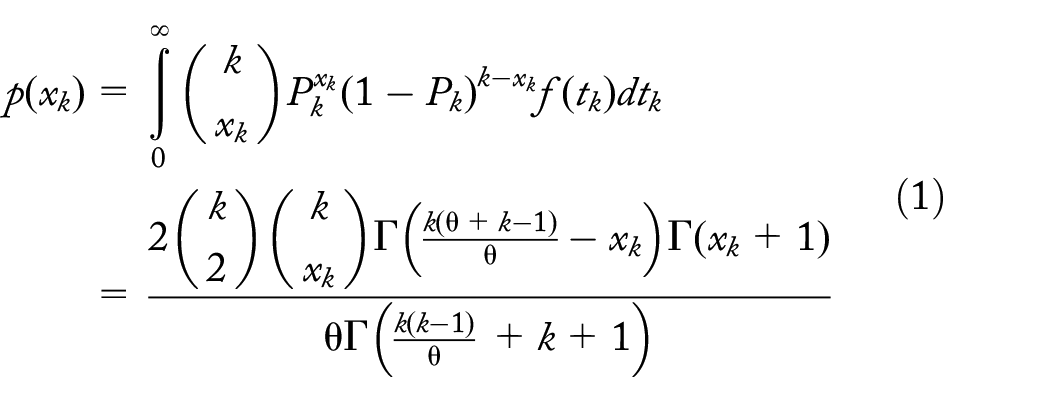
where

and corresponds to the probability of at least 1 mutation prior to a coalescent event at level k (cf. Wakeley 21 ).
Probability
of the
dropouts at level k are unique along their corresponding lines of descent
Define
To calculate the distribution for
Backward in time, 2 events can occur that are relevant to determining the distribution of

for
Generally, for
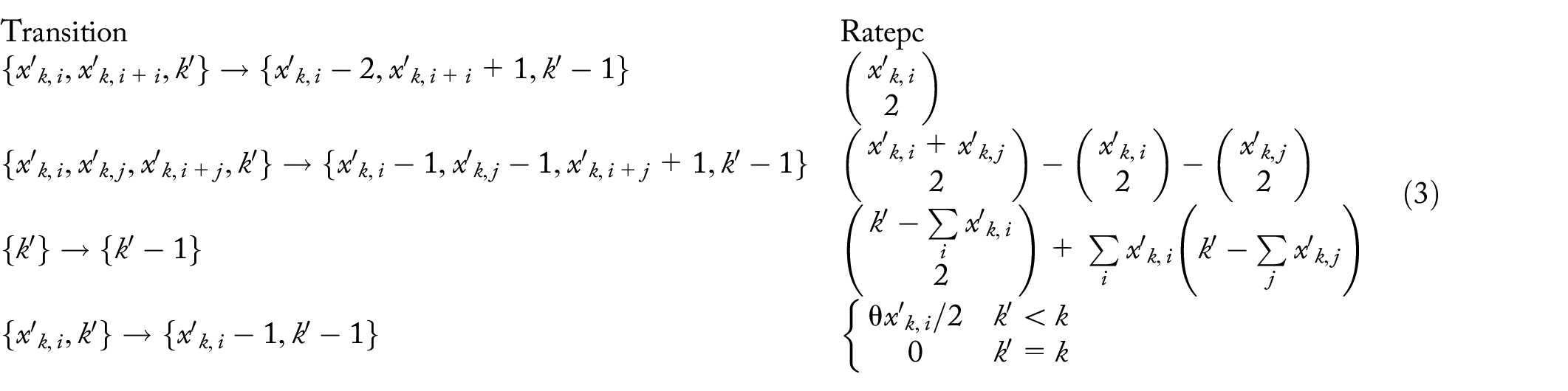
For the set

where
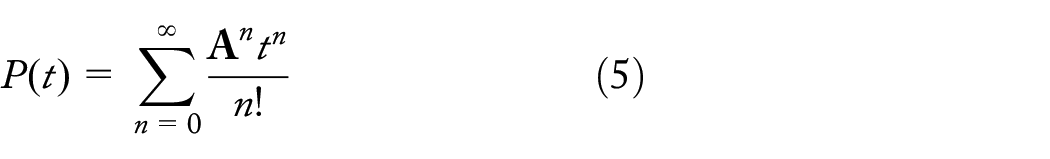
For our purposes, we are interested in the probability at time t that the system is in a state in which
Define

Furthermore, in the limit as

As
For the case
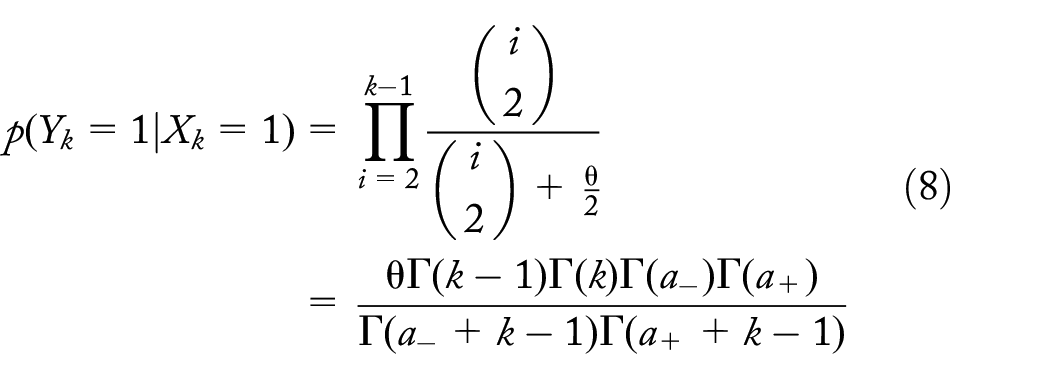
where
Probability of the set of
descendants given
unique dropouts at level k
Define

ways the n individuals can be assigned to k ancestors. Of the k ancestors, we are interested in the number of descendants of each of the

such that the probability of the set

Probability of the set of
descendants of unique dropouts at level k
From the law of conditional probability, the probability of the set of
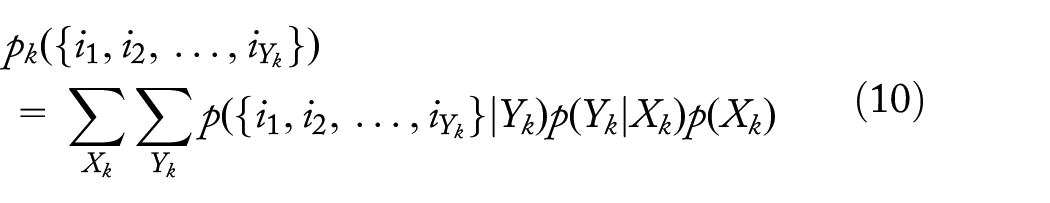
For sufficiently small

For the purposes of metagenomics or eDNA studies, we may be interested in the probability

Distribution of pairwise differences with dropouts
The presence of dropouts may affect the distribution of pairwise differences for a given species. For sufficiently small
Single unique dropout at level 2 (Y2 = 1)
A dropout generates a subtree of
For a subtree of size
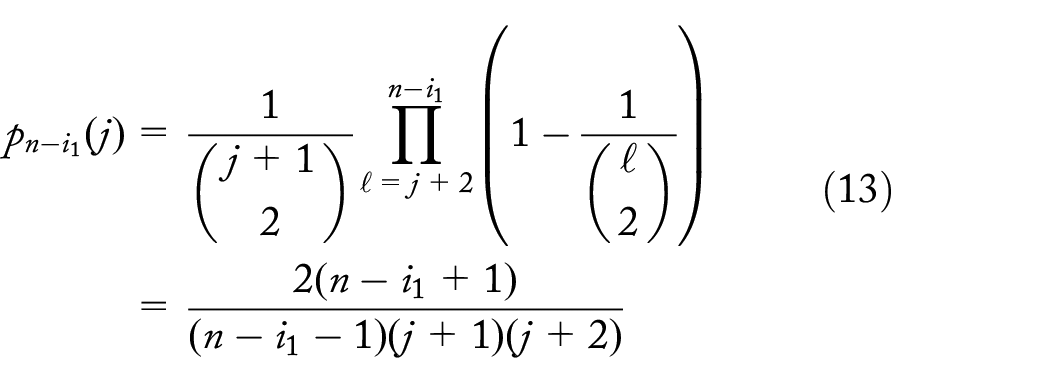
Wiuf and Donnelly
22
derived the probability that, given a subtree first enters level j, the entire sample is at level k
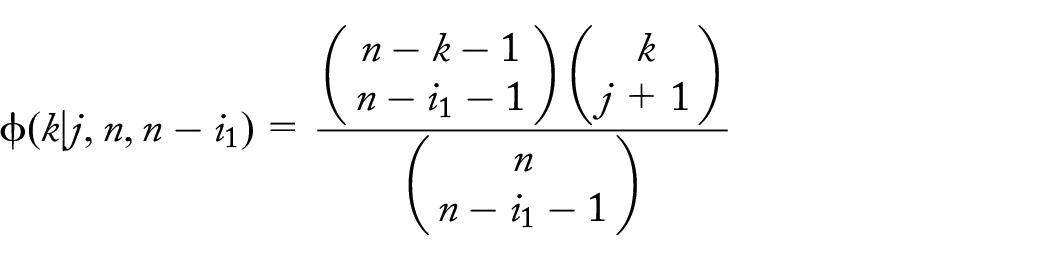
and where here
Given k, the distribution of coalescence times

and can be calculated directed from the sum of exponentially distributed random variables.21,24 Note further that i is indexed initially with
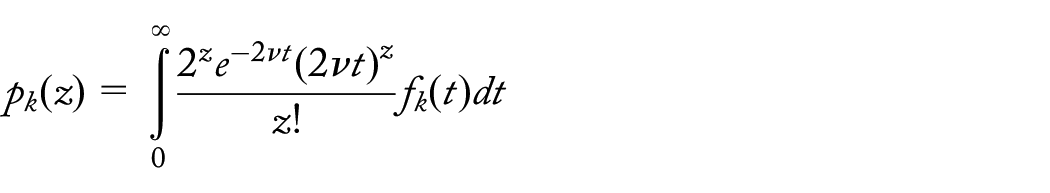
assuming that mutations are Poisson distributed along branch sections and the rate of mutation causing pairwise differences is

Single unique dropout at level 3 (Y3=1)
Here we assume that the dropout mutation occurred such that the 2 clades of sequenced individuals coalesce when

A coalescent tree when
Let
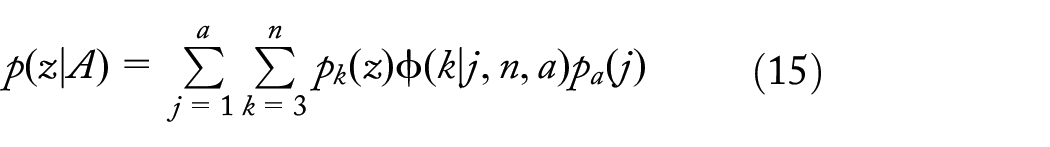
which differs from
When one sequenced individual is in clade A and the other in clade B

and together

The effect of dropouts on coalescence times of sequenced individuals
The previous section demonstrated that dropouts affect the distribution of pairwise differences relative to when there are no dropouts. The difference in the distribution of pairwise difference comes about because of differences in coalescence times, which in turn reflects the
Single unique dropout at level 2 (Y2 = 1)
For

The expected time between the

where

from the standard coalescent.
Single unique dropout at level 3 (Y3 = 1)
As before, we assume that the sequenced individuals are in subclades A and B, as in Figure 3. The numbers of sequenced individuals in subclades A and B are a and b, respectively. We could use a similar approach as in section “Distribution of pairwise differences with dropouts” and condition on each subclade. This conditioning is justified when considering 2 sequenced lineages because either the 2 sequenced lineages occur within the same subclade with
A simple argument indicates that the coalescence times are expected to be distorted with a dropout mutation versus no dropouts. For example, consider a period in the history of the sample in which there are a lineages in clade A and b lineages in clade B, and correspondingly

for

compared with what is expected for an observed sample size of
Neutrality at the primer sites and background selection at the amplified locus
If deleterious mutations occur at rate U and have multiplicative effects on fitness, each with effect s, then the expected frequency of individuals with j deleterious mutations
In principle, we could just scale time as
For a genotype in the jth class, the expected time to reach the 0th class is
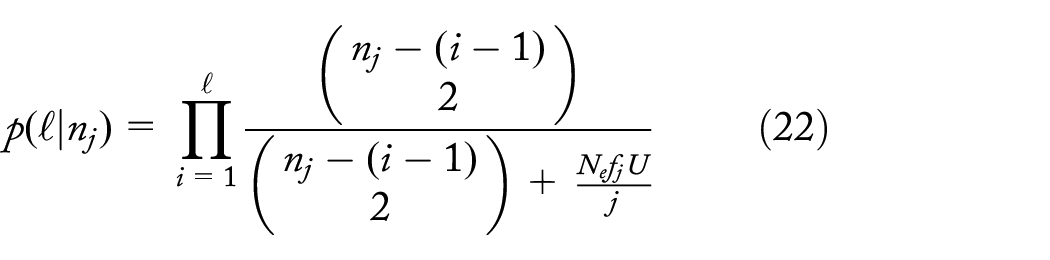
where

where
Once we have
Conditional distribution of pairwise differences with dropouts
Single unique dropout at level 2
With background selection, the corresponding expressions for
For 2 sequences in the same class

For 2 sequences in different classes (

and once in the

Together, the probability of no pairwise difference
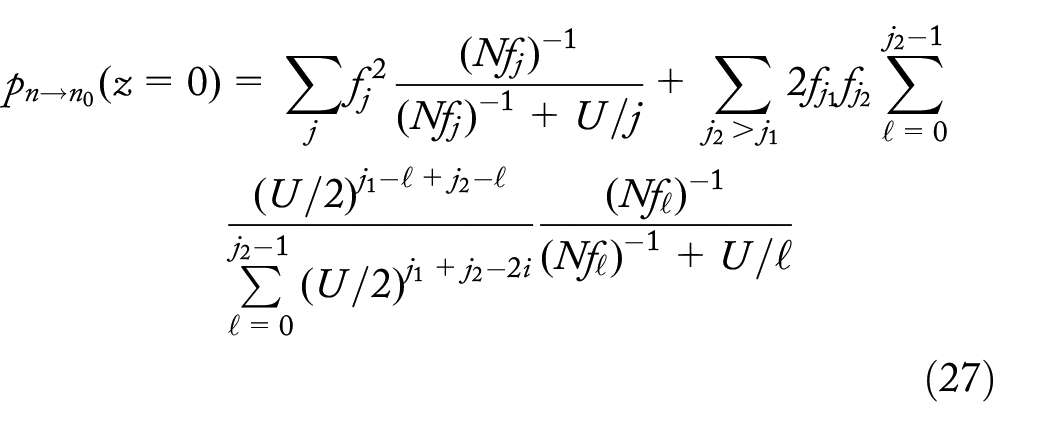
After entering the 0-class, pairwise differences are expected to behave as
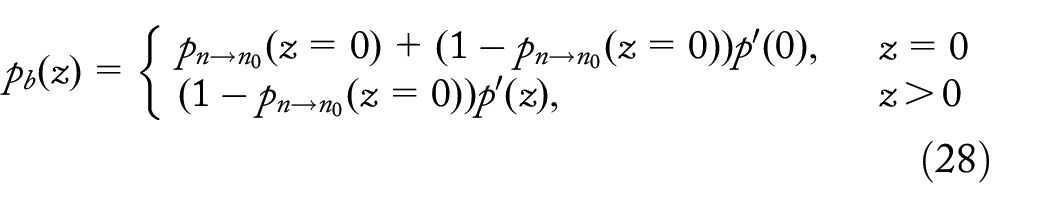
Single unique dropout at level 3 (Y3 = 1)
Following similar logic as for

where again

and

noting that the 2 sequences cannot coalesce during the fast time period when sampled in clades A and B.
The effect of dropouts on coalescence times of sequenced individuals
Once the sample reaches the 0th class mutationally, we expect the effects of dropouts on coalescence times to be the same as the neutral case, except for
Analysis and Results
Expected values for
In the context of dropout mutations,
Under neutrality
Probability of
branches with dropouts at level k,
Figure 4A plots
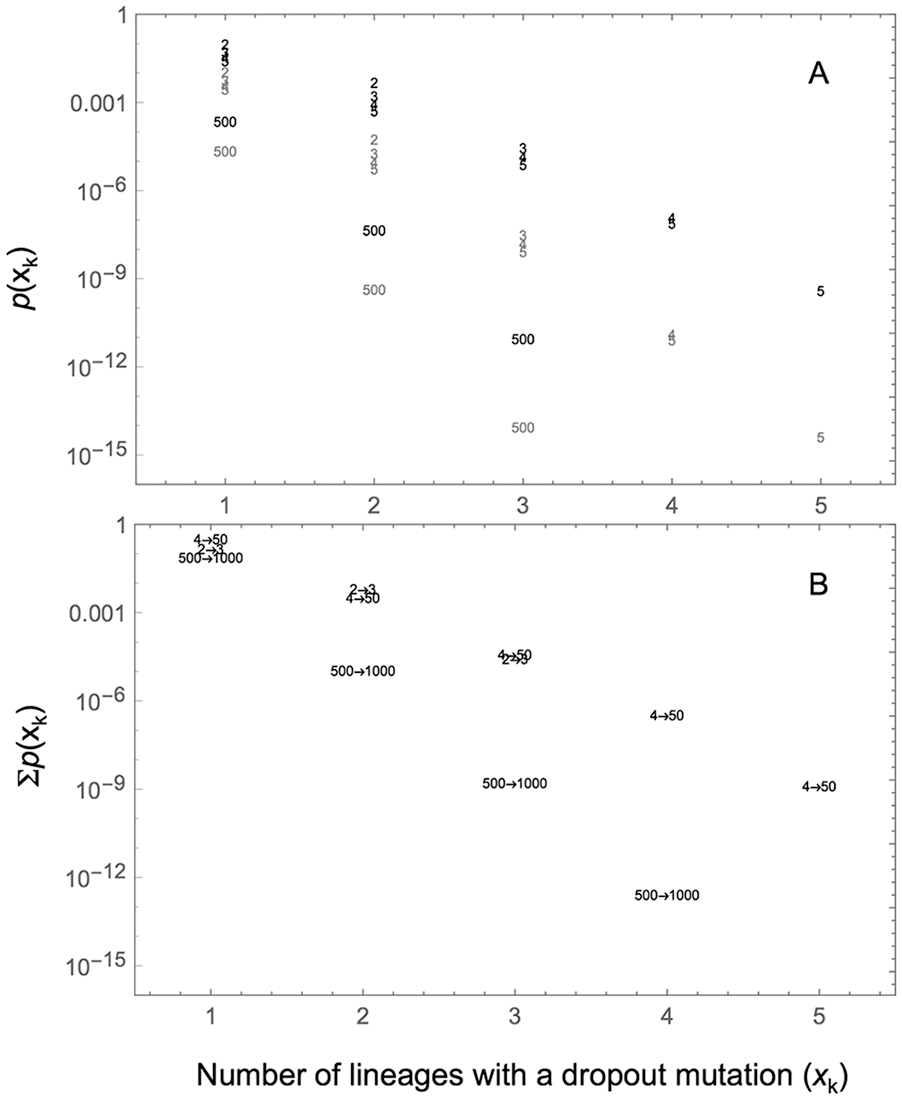
(A) The probability of the number of lineages with a dropout mutation
Probability
of the
dropouts at level k are unique along their corresponding lines of descent,
and
Expressions for

The combined probability of 1 dropout mutation at level k and it being unique
Although single dropouts occur with the highest probability, to illustrate the utility of

and whereas

and, numerically, for

indicating that there is a high probability given 2 dropout mutations at level
Probability of d or more descendent lineages of a unique dropout mutation at level k,
There is a fairly good chance of a unique dropout at level 2 and that it leads to the loss of at least 100 individuals (~8%; Figure 6) for

As
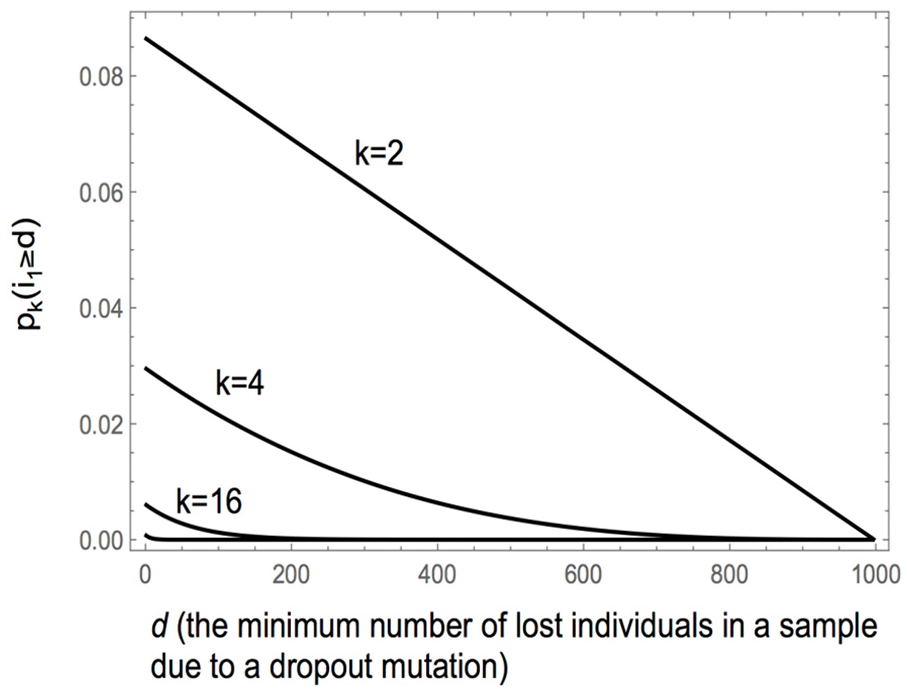
The probability of a dropout out at level k leading to the loss of d or more individuals in a sample

The probability of 2 dropout mutations at level
Distribution of pairwise differences with dropouts and ratio of coalescent times between adjacent nodes
The distribution of pairwise differences is distorted when there is a single dropout at level 2 (Figure 8A). When the number of sequenced individuals is small relative to the entire sample, the distribution of pairwise difference is shifted toward smaller values because sequenced individuals must coalesce before the dropout, which shortens coalesce times. For a moderate to large number of sequenced individuals relative to the entire sample, the distribution can have a mode, unlike the case without dropouts.

The distribution of pairwise differences with (A) 1 dropout at level 2 leading to a single clade of sequenced individuals and (B) 1 dropout mutation at level 3, leading to 2 clades of sequenced individuals with the topology shown in Figure 3. The gray curve shows the distribution in the absence of dropouts. The parameters are
With a dropout leading to 2 clades of sequenced individuals, such that
The ratio of coalescent times between adjacent nodes indicates that it is lower at levels near the base of the tree in the presence of a dropout (Figure 9).
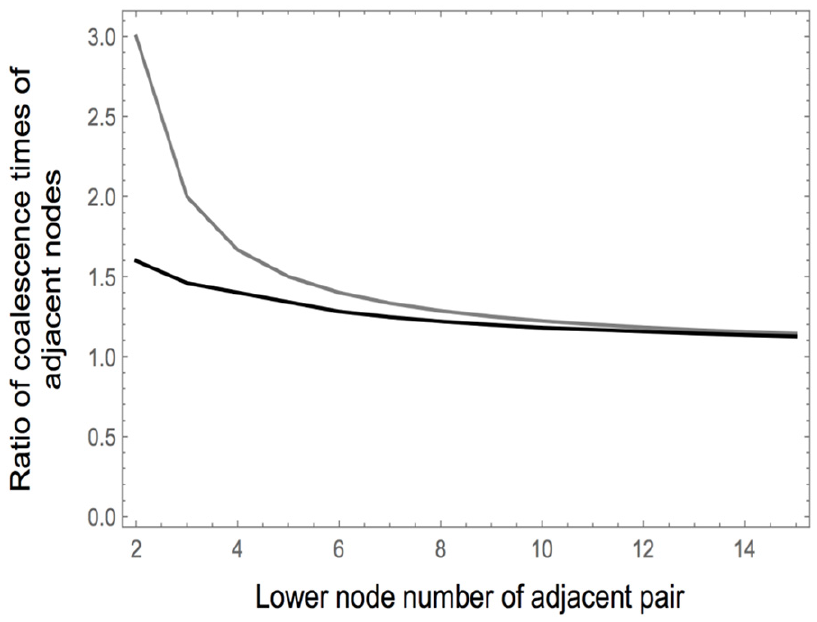
The ratio of coalescence times between adjacent nodes in the absence of a dropout mutation (gray) and in the presence of a dropout at level
With background selection
Expected values for
Background selection is expected to decrease the effective value of
Properties of samples with dropout mutations and background selection
Let us first revisit
Next, consider
The analysis involving
As for large
Discussion
Bacterial and other microbial species are associated with populations of large effective size. This in combination with metagenomic and/or eDNA studies of microbial communities that include thousands of species or OTUs 9 brings about the possibility that one or more of the sampled species or OTUs may be segregating mutations that cause dropouts at PCR primer sites. This article quantified the probability of a segregating dropout mutation at a primer site, as well as the effects of dropouts on the distribution of pairwise differences and coalescence times within a species or OTU.
The analysis indicates that segregating PCR dropout mutations are reasonably common provided that the effective population size of a species is large and/or the dropout mutation rate is reasonably high. Furthermore, if dropout mutations occur, they are expected to occur along basal branch sections in the coalescent tree of a sample. That dropouts are expected to occur along the basal branch section brings about the possibility that a large fraction of sampled individuals go unsequenced due to a dropout mutation within a species. This fraction of individuals that go unsequenced leaves behind a signal in the pattern of pairwise differences, as well as the coalescence times among sequenced individuals. In particular, there is a greater tendency for the distribution of pairwise differences to have a non-zero mode in its distribution, with a mode being particularly pronounced when a dropout mutation gives rise to 2 subclades of sequenced individuals as depicted in Figure 3. Furthermore, a dropout mutation is expected to distort coalescence times near the base of a coalescent tree, such that coalescence times are more equal in value relative to when dropouts are absent. The distortion of coalescence times is independent of n (the true sample size) in the context of equation (21). In other contexts, such as pairwise differences, the effect of dropouts is qualitatively the same across n. Here, inference approaches could integrate across possible values of n to assess whether a distribution is consistent with the presence of one or more dropout mutations.
When a dropout mutation gives rise to 2 clades of sequenced individuals as in Figure 3, the coalescent process for these 2 clades is structured such that individuals between clades cannot coalesce until after the dropout mutation (backwards in time). Qualitatively, the distribution of pairwise differences is similar to that when there is population structure, 25 in which the distribution is a combination of small pairwise differences between individuals within the same clade and large pairwise differences between individuals in different clades. When a single dropout mutation occurs at level 2 in a coalescent tree, its effect on the distribution of pairwise differences is less pronounced than a level 3 dropout mutation that gives rise to 2 clades, such that there is a tendency for the distribution to have a non-zero mode.
A future direction for research is to develop statistical techniques to assess whether a metagenomic or eDNA sample has segregating dropout mutations. Assessing whether a dropout mutation occurs in a single species will be challenging because the pattern that is generated is similar to population structure 26 or changes in population size. 31 Furthermore, the coalescence process in a panmictic population of constant size gives rise to a highly diverse set of distributions of pairwise differences and coalesce times for a given sample size. It may be more promising to first develop methods that seek to assess whether there is a signal of dropouts at the population level across a clade of species or OTUs in a metagenomic study. A consistent signal of dropouts within a clade may lead to greater confidence in the presence of dropouts. A clade of species or OTUs may be predisposed to dropout mutations due to prior substitutions at the primer sites in the ancestor to the clade. We think that the EMBL Metagenomics database 9 provides an ideal resource to begin to assess the potential occurrence of PCR dropout mutations. The database consists of thousands of studies and often within a study there are multiple samples from a site, each of which is sequenced using an amplicon approach. Having multiple samples from a site would allow for the assessment of whether a signature of PCR dropouts, perhaps across species or OTUs within a clade, is replicated across samples. If it is, then this would be evidence against the random occurrence of a pattern that is similar to predictions made in this study.
A consistent signal across samples could nevertheless be due to population structure or changes in population size, instead of PCR dropouts. Although qualitatively there are similarities in patterns of DNA sequence variation, quantitatively there are likely differences that can distinguish PCR dropouts from the population structure and changes in population size. For example, polymorphisms within clades of a coalescent tree are expected to be at different frequencies geographically because of partial isolation, drift, and the history of mutation. In contrast at a single geographic site and with no population structure at a larger spatial scale, we would expect more equal frequencies of polymorphisms, on average, across clades of a coalescent tree in the presence of PCR dropouts. Population growth may be distinguished from PCR dropouts in that recent growth is expected to increase the number of rare polymorphisms in a population, in contrast to PCR dropouts. Overall, the detection of PCR dropouts will likely be quantitative and involve the development of a model-based inference method with the processes of PCR dropout mutations, population structure, and changes in population size co-occurring. Assessment of the occurrence of PCR dropouts would then be probabilistic, whereby the likelihood or posterior probability of occurrence of a set of PCR dropouts would be weighed in combination with parameters that model population structure and demography.
The detection of the presence of dropout mutations may be useful in metagenomic studies that seek to not only identify taxon-level diversity, but also the abundances of taxa in a community. 32 If a taxon has a signature of dropout mutations, then its abundance estimate may be lower than its true value.
Regarding existing methods that use next-generation sequencing approaches to the estimate nucleotide diversity14,15 and phylogenetic structure, 17 our results indicate that PCR dropouts are a factor of consideration. In the case of nucleotide diversity, dropouts give rise to patterns of DNA sequence variation similar to population structure or changes in population size (see above) and therefore are expected to affect estimates of nucleotide diversity. The process of PCR dropout mutations could be added to coalescent-based estimators of population-level statistics, particularly in the context of the use of next-generation sequencing and environmental samples. Likewise, when inferring the phylogenetic structure of environmental samples that use primer-based sequencing, the addition of the process of mutation at primer sites may be warranted, as these mutations may lead to DNA sequence patterns that suggest demographic processes such as population structure and growth. Similar to the previous discussion of detecting PCR dropouts, the incorporation of dropouts to estimators of statistics and phylogenetic structure will likely involve adding the dropout process to model-based approaches, which would then affect the likelihood or posterior distributions of focal statistics or histories.
This article focused on properties of segregating dropout mutations within a species. The development of more powerful methods to detect dropouts will likely involve combining within-species and between-species approaches. In the context of between-species approaches, a promising theoretical framework is Hey’s 14 model of cladogenesis and its potential generalization. In this article, we showed that dropout mutations affect the shape of coalescent trees and it is likely that they also affect the shape of phylogenetic trees. Properties of tree shape at the phylogenetic level under different diversification processes has been a topic of ongoing research, with detectable differences in diversification processes arising from differences in internode lengths in a phylogenetic tree. 33 Differences in internode lengths phylogenetically are analogous to the effect of dropouts on the ratio of coalescence times. A strong signal for the presence of dropouts would be a clade with a distorted tree shape at the species level combined with distorted coalescence times within species within the clade. Of course, this type of inference requires segregating dropouts within species and polymorphism for dropouts at the clade level, whereas a more common pattern may be that an entire clade is not amplified by PCR due to ancestral dropout mutations. Furthermore, it is important to note that expectations about where dropout mutations occur genealogically at the population or species level using the coalescent may not hold at higher taxonomic levels due to differences in the expected distribution of relative internode lengths in a phylogenetic versus coalescent model.
Regarding sequencing methods, a solution to the occurrence of PCR dropout mutations is to use complementary non-overlapping primer sets that both amplify the same genetic locus. 34 Using this approach, a locus in a species’ DNA will be amplified even if there are dropout mutations at a primer binding site. Nevertheless, this approach is not always used and there exist a large number of datasets that did not use complementary non-overlapping primer sets (see Mitchell et al). 9 For these studies, approaches to assess coverage would be useful.
This article is a starting point to derive expectations for the effects of PCR efficiencies on the inference of taxon diversity and within-taxon genetic diversity in other contexts. For example, a common approach to biodiversity assessment is DNA metabarcoding. 35 DNA metabarcoding is often applied to organisms with smaller effective population sizes than prokaryotes, such that the population-level rate of mutation at primer sites is lower. Accordingly, PCR dropout mutations are expected to be less common in these types of studies, but other factors such as differential PCR inhibition or amplification bias may result in the dropout of individual sequences. 36 Generally, accounting for PCR efficiency may be important in studies that seek to infer population-level genetic diversity from metabarcode data. 37
Coalescent theory has been applied in metagenomic studies previously. For example, O’Brien et al 17 derived a coalescent-based approach to infer the phylogenetic relationships of species from a metagenomic sample, and Bittner et al 38 and Liberles et al 39 provide a broad overview for integrating coalescent approaches into metagenomic studies. Furthermore, Johnson and Slatkin 40 presented a coalescent-based approach to detect recombination in a metagenomic sample. Our article suggests that coalescent theory may allow for the detection of PCR primer dropouts in metagenomic studies.
Conclusions
Primer coverage in microbial metagenomic and eDNA studies is a key uncertainty. In this article, we derived coalescent-based expectations for the pattern of DNA sequence variation within a species in the presence of segregating PCR dropout mutations, where a dropout mutation completely blocks PCR amplification. Dropout mutations can alter coalescence times and the distribution of pairwise differences, which may form the basis of statistical techniques to detect or account for reduced primer coverage.
Footnotes
Acknowledgements
I wish to thank C Somerville for her assistance developing this line of research and two reviewers for their comments that improved this work, as well as the Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant program for their support.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the NSERC (Canada) - Discovery program.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
CKG conceived and derived the model, performed its analysis and wrote the paper.