
Abstract
Currently, although many successful bioinformatics efforts have been reported in the epitranscriptomics field for N6-methyladenosine (m6A) site identification, none is focused on the substrate specificity of different m6A-related enzymes, ie, the methyltransferases (writers) and demethylases (erasers). In this work, to untangle the target specificity and the regulatory functions of different RNA m6A writers (METTL3-METT14 and METTL16) and erasers (ALKBH5 and FTO), we extracted 49 genomic features along with the conventional sequence features and used the machine learning approach of random forest to predict their epitranscriptome substrates. Our method achieved reasonable performance on both the writer target prediction (as high as 0.918) and the eraser target prediction (as high as 0.888) in a 5-fold cross-validation, and results of the gene ontology analysis of their preferential targets further revealed the functional relevance of different RNA methylation writers and erasers.
Introduction
Posttranscriptional RNA modifications are important mechanisms that act on all kinds of RNAs, leading to their increased structural and functional diversity. 1 There are at least 100 kinds of RNA modifications, 2 among which N6-methyladenosine (m6A) is currently the most prevalent and intensively studied due to its wide impacts. 3 It regulates many essential biological processes including neuronal differentiation, obesity, and messenger RNA (mRNA) stability.4-6 The m6A RNA methylation is a reversible mark, which is deposited by methyltransferases (or the writers), including METTL3 (methyltransferase-like 3), METTL14 (methyltransferase-like 14), METTL16 (methyltransferase-like 16), and so on, and is removed by demethylases (or the erasers), including FTO (fat mass and obesity–associated protein) and ALKBH5 (ALKB homolog 5).
The writers of RNA m6A modification are protein complexes containing catalytic components METTL3, METTL14, and METTL16, which all have the class I methyltransferase domain. METTL3 is the first identified m6A relevant methyltransferase that has S-adenosylmethionine (SAM)-binding activity. 7 Afterward, METTL14 was discovered as the second methyltransferase that has a methyltransferase domain sharing 22% sequence identity with METTL3. 8 While individual METTL3 or METTL14 exhibits comparably weak catalytic activity in vitro, the METTL3-METTL14 complex has higher catalytic capacity.9,10 In addition, the crystal structure of METTL3-METTL4 complex suggested that only METTL3 binds with SAM and METTL14 plays a structural role for substrate recognition.8,11 Thus, the heterodimeric METTL3-METTL14 complex was considered a catalytic domain of m6A methyltransferase. Recently, METTL16 has been identified as another catalytically active m6A mRNA methyltransferase. 12 The METTL16 is similar to METTL3 in structure, but has some unique elements, such as unique αB helix in the Rossmann fold. 13 In addition, these 2 catalytically active m6A mRNA methyltransferases have different roles to play in biological processes. For example, the METTL14 and METTL3 modulate cell cycle progression of cortical neural progenitor cells 14 and depletion of METTL3 or METTL14 promotes tumor progression by enhancing the growth of glioblastoma stem cells. 15 METTL16 can recognize hairpin and methylated adenosine in the U6 snRNA, which regulates the expression of MAT2A. 16
FTO and ALKBH5 are 2 currently identified m6A-specific RNA demethylases (erasers).4,17 Although FTO is able to act as a demethylase on another substrate, N3-methylthymidine (m3T), its efficiency is much lower than working on m6A substrates.18,19 Although both FTO and ALKBH5 can target specifically RNA m6A,19,20 the 2 differ significantly on many levels. For example, at the molecular level, FTO has an amino-terminal AlkB-like domain, a carboxy-terminal domain with a novel fold, and an extra loop that covers on one side of conserved jelly-roll motif. 21 ALKBH5 is a member of the 2-oxoglutarate (2OG) and ferrous iron-dependent nucleic acid oxygenase (NAOX) families, it has a double-stranded β-helix core fold and the active metal site is coordinated by an HXD. . .H motif along with 3 water molecules. 22 In addition, their reaction pathways seem to be different: m6A is directly converted by ALKBH5 to adenosine; 2 intermediates, N6-hydroxymethyladenosine (hm6A) and N6-formyladenosine (fm6A), are observed during demethylation of m6A sites by FTO.23,24 FTO and ALKBH5 also play different roles in terms of physiological functions, one is associated with obesity 4 and the other is thought to participate in the formation of sperm. 25 Moreover, FTO is mainly expressed in the brain, 26 in contrast to ALKBH5, which is found in most tissues, particularly in the testes. 17
Therefore, according to these studies, 2 kinds of catalytically active m6A mRNA methyltransferases and 2 demethylases exist that have distinct structures and participate in different biological functions. It would be very interesting to know what the preferential target sites of METTL3-METTL14 complex, METTL16, FTO, and ALKBH5 are and their downstream biological processes. Experimental approaches are effective for testing their functional relevance under a specific experimental condition, such as using different cell lines or testing different treatments. Due to limited detectability, it is not possible to detect target sites on very lowly expressed genes, which is the intrinsic limitation of wet lab–based approach, such as ParCLIP. To unveil comprehensively the epitranscriptome-wide targets of RNA m6A, we considered using computational approaches.
Currently, the field of bioinformatics has seen the rapid development of new methods and their wide applications in RNA epigenetics. The mammalian m6A short consensus motif RRACH (where R = A or G; H = A, C, or U) has not been characterized until 2012, when the next-generation sequencing techniques called m6A-seq or MeRIP-seq (methylated RNA immunoprecipitation sequencing)27-29 emerged. Thereupon, RMBase and MeTDB have been developed into v2.0, which now can provide millions of m6A sites in many different species, such as, human, mouse, yeast, and fly.30,31
Meanwhile, many successful computational studies have been devised on m6A site prediction, such as SRAMP, MethyRNA, and RNAMethPre.32-34 Although there are many good precedents in m6A site prediction and deposition (database development), there has been no effort made in the substrate prediction of m6A enzymes. We, therefore, devised a computational tool to study the target specificity of m6A enzymes. In this study, the predictors were built using the random forest (RF) approach to distinguish the target specificity of the m6A writers (METTL3-METTL14 complex and METTL16) and the erasers (FTO and ALKBH5), respectively. Although the sequence-derived features were widely used in m6A site prediction33,35 and generated reasonably good results, we included additional genome-derived features and achieved substantial improvement in performance.
Matrerials and Methods
The m6A sites
The transcriptome-wide m6A sites were extracted from the WHISTLE web server, 36 which used multiple genomic and sequence features to predict the entire epitranscriptome and achieved substantial improvement compared with existing approaches. Please note that all these m6A sites were originally collected from wet lab experiments 30 and simultaneously supported by the WHISTLE prediction with high confidence. We considered the m6A sites with probability greater than .6, .7, .8, and .9, which are corresponding to 4 data sets of 98 095, 75 720, 52 687, and 27 646 RNA methylation sites, respectively. In this study, 4 different sets of data were extracted for further analysis. This is because they correspond to different coverage and reliability. A larger set has better coverage of the m6A epitranscriptome, but may also contain more false m6A sites that can affect prediction performance. The training data is provided in Supplementary Table 1.
Target sites of the enzymes
The ground truth targets were identified using perturbation experiment, eg, the hypomethylated sites after the knock down of a methyltransferase identified from MeRIP-seq data. Specifically, the raw data were retrieved from GEO (Gene Expression Omnibus; see Table 1), and the FASTQ files were aligned to the reference genome hg19 using hisat2 42 with default settings. The resulting SAM files were then converted to BAM files using samtools with the quality filter –q 30 and the FLAG filter –F 2820. Following that, the number of reads aligned to each individual RNA methylation sites were counted as fragments in R using GenomicAlignment package. 43 For each experiment with regulator perturbation, differential methylation analysis was conducted by DESeq2 44 using the interactive generalized linear model (GLM) design of ~ IP*Treatment, while IP is the indicator vector for the samples being IP, and Treatment is the indicator vector for the samples treated with regulator perturbation. The m6A sites with the Wald test fdr < 0.05 and the interactive coefficient <0 (>0 for the sample gsc11-ALKBH5-) are treated as the target sites of the regulator. The shared target sites of multiple enzymes, ie, (FTO and ALKBH5) or (M3/M14 vs M16) are considered with ambiguous association and thus excluded from our analysis.
GEO data sets used to identify ground truth target sites.

Abbreviations: ALKBH5, ALKB homolog 5; FTO, fat mass and obesity–associated protein; GEO, Gene Expression Omnibus; METTL3, methyltransferase-like 3; METTL14, methyltransferase-like 14; METTL16, methyltransferase-like 16; SRA, Sequence Read Archive.
Feature encoding scheme and selection
Sequence-derived features
The nucleotide encoding method according to chemical properties was suggested by Bari et al. 45 In the MethyRNA 32 and M6Apred, 46 this encoding method was applied in the generation of sequence-derived features and achieved good accuracy in the m6A site prediction. In this project, we followed this idea of chemical encoding method to generate sequence-derived features. Specifically, 3 chemical properties of the nucleotides were used to classify adenine (A), cytosine (C), guanine (G), and uracil (U). The first property is ring structures: A and G have 2 ring structures, whereas C and U have only 1 ring. The second property is functional groups. A and C contain amino group, whereas G and U contain the keto group. The third property is the number of hydrogen bonds formed. A and U can form 2 hydrogen bonds during hybridization, whereas G and C can form 3 hydrogen bonds. Based on the 3 structural chemical properties defined above, the ith nucleotide from sequence can be encoded by a vector:
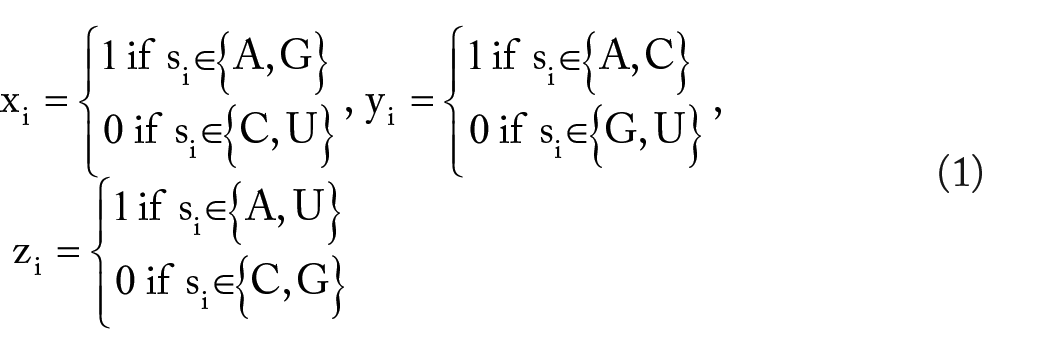
Thus, A can be marked as (1, 1, 1), C can be marked as (0, 1, 0), G can be marked as (1, 0, 0), and U can be marked as (0, 0, 1). In addition, a feature of the accumulative nucleotide frequency is calculated for each nucleotide position in the sequence. The density of ith nucleotide is defined as the sum of all the instances of the ith nucleotide before the
Genome-derived features
Although the sequence-based features were widely used in the prediction of RNA modification sites, there are potentially other features that can be used. 47 The genomic features have been shown in the WHISTLE project to be effective in the m6A site prediction. A total of 47 genome-derived features were considered for this project (see Table 2). Specifically, genomic features 1 to 17 specify the locations of adenosine sites within the transcript region and their topological properties as dummy variables. To generate features in this category, we used the transcript annotations of hg19 human genome assembly and the GenomicFeatures R/Bioconductor package. 43 Genomic features 18 to 21 define the relative positions of adenosine sites within transcript region, which is calculated based on the distance from the methylated adenine to the 5′ end divided by the total width of the region; the position features are set to 0 if the adenosine sites do not belong to the region. The values of features 22 to 26 are lengths of the transcript region containing the methylated site; if the sites do not belong to the region, the features are set as 0 also. The evolutionary conservation score of the methylated adenosine sites and its flanking regions was measured in features 27 to 30 with 2 metrics of nucleotide conservation: PhastCons score and the fitness consequence scores. Features 31 and 32 represent the RNA secondary structures of transcripts region containing methylated adenine predicted using RNAfold in Vienna RNA package. Features 33 to 35 represent the attributes of the genes or transcripts containing methylated sites. Features 36 to 40 indicate whether the adenosine sites interact with small noncoding RNA, long noncoding RNA, and microRNA, respectively. Finally, features 41 to 49 indicate whether the methylated sites are located within RNA protein-binding regions. For the above features, to avoid ambiguity caused by transcript isoforms, only the primary (longest) transcript of each gene was kept for the extraction of the transcript subregions. The details for genomic features are summarized in Table 2.
Genomic features used in the analysis.

Abbreviations: ALKBH5, ALKB homolog 5; FTO, fat mass and obesity–associated protein; GEO, Gene Expression Omnibus; METTL3, methyltransferase-like 3; METTL14, methyltransferase-like 14; METTL16, methyltransferase-like 16; SRA, Sequence Read Archive.
Features that are directly related to the prediction are not used to avoid overfitting. For example, the features 42 and 43 were not used for writer target prediction, whereas feature 48 and 49 were not used for eraser target prediction.
Machine learning approach
Machine learning algorithms have been widely used in the field of computational biology. In RNA epigenetics studies, support vector machine (SVM) and RF have been used previously in RNA m6A site prediction,32,33,46 and both achieved good performances. In this project, the RF algorithm from the R randomForest was used to build predictor models.
Performance evaluation
A 5-fold cross-validation was used for assessing the reliability of the method. In the performance evaluation, the sensitivity


where TP, TN, FP, and FN represent true positive, true negative, false positive, and false negative, respectively. In addition, prediction performance under different decision thresholds were measured as the receiver operating characteristic (ROC) curve whose y-axis is sensitivity and x-axis is 1-specificity, the area under ROC curve (AUC) was calculated as the main performance evaluation metrics. In addition, the ACC (overall accuracy) and MCC (Matthews correlation coefficient) were calculated as other indicators to evaluate the reliable of model.
Results and Discussion
Feature selection
Although extensive research in m6A site prediction has demonstrated the effectiveness and reliability of sequence-derived features32,33,54 and genome-derived features, 36 we seek to, for the first time, use these features to predict the target specificity of m6A enzymes. Due to the abundance of the genomic features, we first performed feature selection to identify the genomic features most relevant to our purpose, which is to improve the reliability of the features and the prediction performance, as well as to save computation time and memory. The feature selection was performed on the set of RNA methylation sites with probability greater than .6.
At the beginning, the Perturb method 55 was used to estimate the relative importance of each genomic feature in the target specificity prediction of eraser targets using the R caret package. To illustrate the relative importance of different features clearly, the measurement results are rescaled and ranked (Figure 1A). According to this rank, relevant AUROC (area under the receiver operating characteristics) figures are generated based on the top N features. We can observe that the best performance was achieved with the top 20 features. Thus, only the top 20 features were used in our prediction model for erasers. Similarly, the same treatment was done on the writer target prediction (Figure 1B), where the best performance was achieved with the top 15 features.
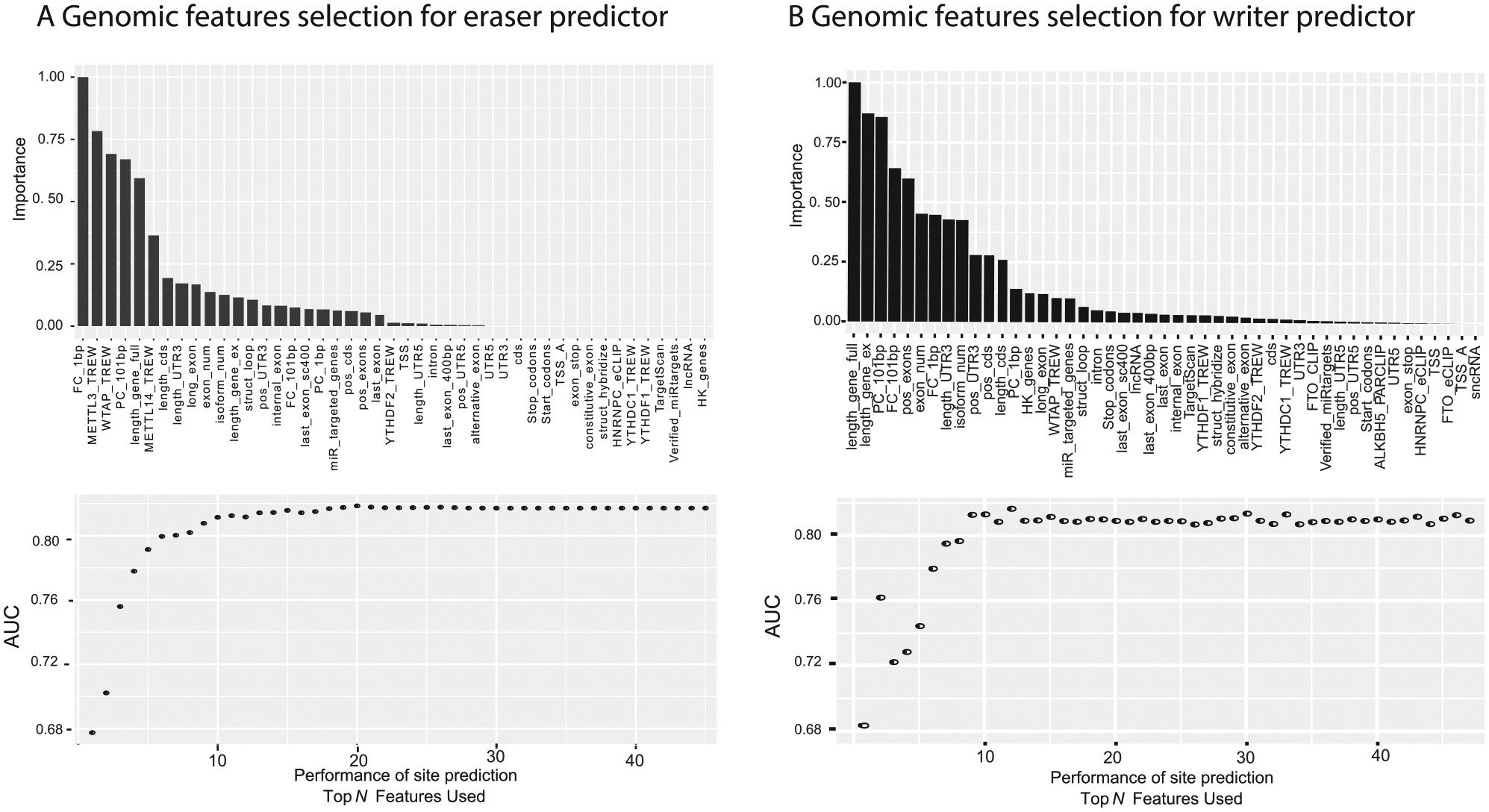
Feature Selection for Predictors. (A) The top 20 genomic features were used for prediction of the targets of erasers, including conservation score, METTL3 targets, etc. (B) The top 15 genomic features were used for prediction of the targets of writers, with the distance to known m6A site as the most important predictive feature, followed by gene length and conservation score.
Predictors based on different set of features
Existing computation models overwhelmingly relied on the sequence features. In our prediction model, while it also incorporates features derived from other genomic annotations (see Table 2), It is important to test whether these features contribute to the prediction performance. For this purpose, a 5-fold cross-validation was conducted on the data set of RNA methylation sites with probability greater than .6, and different types of features were used. As shown in Table 3, sequence features were more effective than sequence features in the target prediction for erasers; but not in the case for writers. However, it is consistent for the best performance to be achieved when both sequence and genomic features were incorporated.
Performance of predictors based on different features.

Abbreviations: ALKBH5, ALKB homolog 5; AUROC, area under the receiver operating characteristics; FTO, fat mass and obesity–associated protein.
This result was achieved on RNA methylation sites with probability greater than .6 with a 5-fold cross-validation.
Performance on different data sets
In next step, we consider expanding the model to test on all the 4 data sets. As shown in Table 4, the 4 different data sets have different coverage of the m6A epitranscriptome; for erasers, the best target prediction performance was achieved on data set 4, which are RNA methylation sites with probability greater than .9, whereas for writers, the best performance was achieved on data set 3, which are corresponding to the RNA methylation sites with probability greater than .8. To compare with other machine learning approaches, the SVM, Naïve Bayes, decision tree, and GLM were applied to build model. The performances for each method are summarized in Table S2 and were evaluated by the sensitivity, specificity, AUROC, ACC, and MCC.
Prediction performance on different data sets (AUROC).

Abbreviations: ALKBH5, ALKB homolog 5; AUROC, area under the receiver operating characteristics; FTO, fat mass and obesity–associated protein.
Four data sets were considered, corresponding to the experiment-validated RNA methylation sites from RMBase and also supported by WHISTLE prediction with probability greater than .6, .7, .8, and .9, respectively. The detailed performance of 5 different classification predictors (RF, SVM, GLM, Naïve Bayes, and decision tree) is presented in Supplementary Table S2.
Biological functions regulated by different enzymes
There are 3585, 4623, 4742, and 4734 sites identified in data set 1 (see Table 4) under the regulation of METTL3-METTL14 complex, METTL16, FTO, and ALKBH5, respectively, which are located on 2149, 2178, 2375, and 2635 genes. The biological functions of these targets sites were then annotated with gene ontology enrichment analysis using DAVID website. 56 Figure 2 shows the top 10 mostly enriched biological processes. We can see that different biological processes were enriched in different enzymes. For example, FTO is associated with cell-cell adhesion (5.473E−13), mRNA splicing (2.212E−12), viral process (4.31E−09), whereas the target sites of ALKBH5 are more related to Golgi organization (4.13E−09) and DNA-templated transcription (4.14E−14). METTL16 targets are enriched with genes related to endoplasmic reticulum–associated misfolded protein catabolic process (9.07E−06), regulation of cell cycle (1.15E−04), apoptotic process (6.70E−04) and protein ubiquitination (9.99E−05), whereas METTL3-METTL14 complex preferentially target to genes associated to cell-cell adhesion (2.86E−07), cell division (4.08E−07), and G2/M transition of mitotic cell cycle (3.03E−06). Please see Table S3 for the complete gene ontology enrichment analysis result.
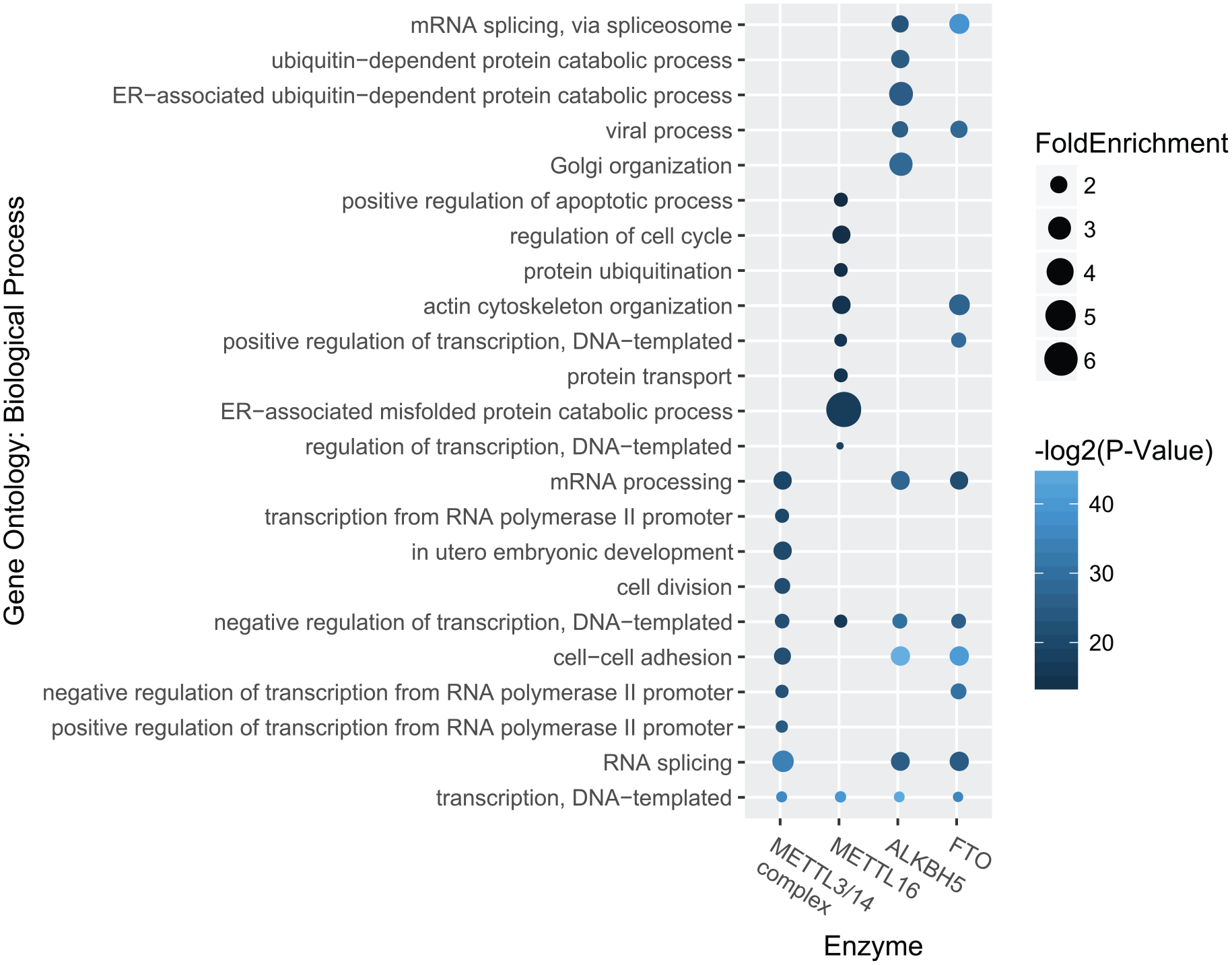
Biological processes enriched in targets of m6A enzymes. Distinct biological processes are enriched in the predicted target sites of different enzymes. Figure shows the top 10 most statistically enriched biological processes associated with the targets of different m6A enzymes.
Discussion and Conclusions
Recent progress in RNA modification bioinformatics enabled the precise detection, accurate quantification, differential analysis, and function annotation of m6A RNA methylation sites in base resolution. RMBase and MetDB collected experimentally validated m6A sites in multiple species and revealed their potential regulatory functions.30,31 The exomePeak57,58 was developed based on Przyborowski and Wilenski’s59,60 method for m6A site detection and differential methylation analysis from MeRIP-seq data. The computational prediction of m6A modification sites in different species performed in the works iRNA(m6A)-PseDNC, 61 iRNA-Methyl, 62 m6Apred, 46 RFAthM6A, 63 and BERMP 64 based on machine learning or deep learning approaches. The potential disease relevance and single-nucleotide polymorphism association of m6A modification were revealed by the m6Avar 65 and m6ASNP 66 by examining whether a disease mutation can alter the potential of RNA methylation status. Meanwhile, complex network method was used in m6Acomet, 67 m6A-Driver, 68 Deepm6A, 69 DRUM, 70 and FunDMDeep-m6A 71 to study the regulatory functions and predict the disease association of m6A RNA modification.
Here, we have proposed a computational approach for the prediction of the target sites of m6A enzymes. The computational model proposed relies on 49 genomic features as well as the conventional sequence features. With a model selection step, we showed with a 5-fold cross-validation that the proposed approach achieved relatively good performance in the target prediction for the writers (AUC: 0.918) and erasers (AUC: 0.888). The following gene ontology analysis unveiled the epitranscriptome functional relevance of these enzymes.
The proposed approach suffers from the following limitations. (1) The ground truth target sites were identified from perturbation experiment, in which a target site of a methyltransferase is defined as those whose methylation level decreases when the methyltransferase was knocked down. Obviously, the decrease in methylation level may not be due to direct target but because of a secondary effect. For this reason, the ground truth data can be further improved. (2) The features incorporated in the prediction model can be further increased. Although a total of 49 genomic features have been incorporated in our prediction model, the set can be expanded by including, eg, features related to lncRNA, repeat region. Increased feature set can often lead to improved performance. (3) We considered here only a binary classification, which emphasizes the target specificity of different enzymes. However, in practice, it is possible that there are a large number of RNA methylation sites that are simultaneously targeted by both m6A writers (or both m6A erasers) considered in this work. In addition, there are likely to be unknown methyltransferases or demethylases to be discovered and thus are not considered in the prediction models. This would be a difficult question to solve. (4) A better computation method may be used. We used here RF, which is a classic method. Recent development in artificial intelligence, especially deep learning–related approach may achieve better performance.
Supplemental Material
Supplement_Table_1 – Supplemental material for Predict Epitranscriptome Targets and Regulatory Functions of N6-Methyladenosine (m6A) Writers and Erasers
Supplemental material, Supplement_Table_1 for Predict Epitranscriptome Targets and Regulatory Functions of N6-Methyladenosine (m6A) Writers and Erasers by Yiyou Song, Qingru Xu, Zhen Wei, Di Zhen, Jionglong Su, Kunqi Chen and Jia Meng in Evolutionary Bioinformatics
Supplemental Material
Supplement_Table_2 – Supplemental material for Predict Epitranscriptome Targets and Regulatory Functions of N6-Methyladenosine (m6A) Writers and Erasers
Supplemental material, Supplement_Table_2 for Predict Epitranscriptome Targets and Regulatory Functions of N6-Methyladenosine (m6A) Writers and Erasers by Yiyou Song, Qingru Xu, Zhen Wei, Di Zhen, Jionglong Su, Kunqi Chen and Jia Meng in Evolutionary Bioinformatics
Supplemental Material
Supplement_Table_3 – Supplemental material for Predict Epitranscriptome Targets and Regulatory Functions of N6-Methyladenosine (m6A) Writers and Erasers
Supplemental material, Supplement_Table_3 for Predict Epitranscriptome Targets and Regulatory Functions of N6-Methyladenosine (m6A) Writers and Erasers by Yiyou Song, Qingru Xu, Zhen Wei, Di Zhen, Jionglong Su, Kunqi Chen and Jia Meng in Evolutionary Bioinformatics
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work has been supported by National Natural Science Foundation of China (31671373), Jiangsu University Natural Science Program (16KJB180027), XJTLU Key Program Special Fund (KSF-T-01), and Jiangsu Six Talent Peak Program (XYDXX-118).
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
JM and KC conceived the idea and designed the research; ZW processed the raw data; YS, QX, and DZ performed the prediction analysis; YS and QX drafted the manuscript first. All authors read, critically revised, and approved the final manuscript.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.