
Abstract
Phylogenetic methods unearth evolutionary history when supported by three starting points of reason: (1) the continuity axiom begs the existence of a “model” of evolutionary change, (2) the singularity axiom defines the historical ground plan (phylogeny) in which biological entities (taxa) evolve, and (3) the memory axiom demands identification of biological attributes (characters) with historical information. Axiom consequences are interlinked, making the retrodiction enterprise an endeavor of reciprocal fulfillment. In particular, establishing direction of evolutionary change (character polarization) roots phylogenies and enables testing the existence of historical memory (homology). Unfortunately, rooting phylogenies, especially the “tree of life,” generally follow narratives instead of integrating empirical and theoretical knowledge of retrodictive exploration. This stems mostly from a focus on molecular sequence analysis and uncertainties about rooting methods. Here, we review available rooting criteria, highlighting the need to minimize both ad hoc and auxiliary assumptions, especially argumentative ad hocness. We show that while the outgroup comparison method has been widely adopted, the generality criterion of nesting and additive phylogenetic change embodied in Weston rule offers the most powerful rooting approach. We also propose a change of focus, from phylogenies that describe the evolution of biological systems to those that describe the evolution of parts of those systems. This weakens violation of character independence, helps formalize the generality criterion of rooting, and provides new ways to study the problem of evolution.
Keywords
Introduction
Science requires that choice among theories be decided by evidence, and the effect of an ad hoc hypothesis is precisely to dispose of an observation that otherwise would provide evidence against a theory. If such disposals were allowed freely, there could be no effective connection between theory and observation, and the concept of evidence would be meaningless.
Understanding life requires unearthing its history. Retrodiction, the exploration of the past to predict present and future, represents a most challenging proposition. It demands extracting empirical evidence that is present in extant life, the explanandum (ie, the phenomenon to be explained), while using it appropriately to uncover evolutionary change that happened in the past, the explanans (ie, the explanation of the phenomenon). The challenge increases as we travel deeper in time. For that reason, unearthing biological history involves the development of a tightly integrated theoretical (epistemological) and empirical (analytic) framework. Darwin and Wallace, with background knowledge from many that preceded and followed them (especially Owen, Lankester, and Osborn, who elaborated the concept of “homology”), provided foundations for the former. Hennig 2 and the cladistic school formalized the latter. The epistemological and analytical integration gave rise to phylogenetic systematics, seeding the fields of molecular evolution, network biology, and evolutionary genomics. The modern ideographic (historical and retrodictive) rationale of phylogenetic analysis adheres to the hypothetico-deductive method for overthrowing theories that supports scientific growth.3,4 It also embraces a wide diversity of philosophical and quantitative approaches, some of which have been fiercely debated for half a century. These include the contest between parametric (statistical) and non-parametric (parsimony) views of phylogenetic reconstruction.5,6 Within this background, the systematization of phylogenetic analysis has materialized in the reconstruction of a Tree of Life (ToL), a genealogy that summarizes the origin and evolution of organismal diversity at planetary scale (eg, Hinchliff et al 7 ). This remarkable community effort as well as other explorations in ecology and evolution have been hampered by some important choices taken in the pursuit of the ideographic method. Here, we discuss these shortcomings and review one of the most fundamental problems of retrodiction, defining the “arrow of time” of evolutionary change (borrowing from Eddington entropy-induced asymmetry).
The Basics of Phylogeny Reconstruction
Three starting points of reason support evolutionary thinking (modified from Wiley 8 ; Table 1). These primary axioms are inductive statements of the highest level of universality that apply to the entire history of life, from its origin to the present. Their high explanatory power provides fruitful principles of discovery and helps formalize ideographic research. Axiom 1 (continuity) begs the existence of a “model” describing evolutionary change. Axiom 2 (singularity) defines the ground plan of the historical account (“phylogeny”) and the genealogy of biological entities (“phylogenetic taxa”) unfolding in time. Axiom 3 (memory) requires identification of useful biological attributes (“phylogenetic characters”) carrying sufficient historical information. Characters and phylogenetic taxa embody the data or empirical evidence. As we will now make clear, the tripartite interaction of evolutionary model, phylogeny, and data (character/taxa) is subtle and must occur in ways that enhance retrodictive power through test and corroboration.3,4
The three starting points of reason supporting evolution.

First, the phylogenetic implementation of axiom 1 implies establishing an evolutionary model of change. By definition, a character implies a “transformation series,” a set of possible instantiations of the character, the “character states,” and the set of possible transformations (changes) between those states. For example, an amino acid site in the amino acid sequence of an evolving protein can take the form of a character with 20+ possible character states, with character states transforming into any other state, every time there is an amino acid substitution in a sequence. Thus, character transformations are the actual character state changes transmitting change as the phylogeny unfolds. In other words, characters are transmitted modified or unmodified through the genealogical historical account. By definition, this transmission is conservative from an evolutionary point of view. Because instantaneous change is unidirectional, character transformations must be directional, ie, they must show at least two character states (transformational homologs): one ancestral (a “plesiomorphic” state) and the other derived (an “apomorphic” state). This is necessary to define the arrow of time. We note that imparting directionality to character change, ie, “polarizing” character change, does not necessarily imply change always occurs from the plesiomorphic to the apomorphic state, as reversals are known to occur freely in evolution. 9 In fact, in one extreme case, change can be so dynamic that polarization cannot be imparted onto the model without serious consequences to its validity. For example, there is no current rationale to polarize characters of amino acid sequences. Even if an amino acid or nucleotide is discovered to be ancestral, the mutational dynamics of amino acid substitution in proteins has been so massive (sometimes involving billions of years of evolutionary change 10 ) that proposing an ancestral-descendant relationship is unfeasible on the grounds of mutational saturation alone. Adding to this objection is the fact that models of sequence evolution may not be universally applicable to all organisms, as fast-evolving lineages (eg, Nanoarchaea) and sequence sites have been identified. Conversely, there could be processes of change that resist reversibility. For example, the formation of complex cellular structures (eg, organelles or macromolecular assemblies such as the ATP synthase rotor or the flagellum) involves the establishment of numerous molecular and cellular interactions at many different levels of biological organization. Once these interactions that hold structures together are established in an organismal lineage, their elimination by mutational change can be extremely difficult. The character becomes “canalized” and its loss unlikely (see Camin-Sokal and Dollo optimization below).
Once a model of change is envisioned, a character transformation series that is grounded in biological reality must be implemented. Within a generalized framework of maximum parsimony, this is usually done by first using “character state graphs” (CSGs) to describe character states and how they transform into each other 11 and second by invoking “character state matrices” that make explicit characters state transformation costs. 12 Figure 1 shows important examples, which we will soon discuss. However, within a maximum likelihood framework of phylogenetic reconstruction, character change probabilities are made explicit in the parametric model. 14 This usually takes the form of a Q matrix, a table of instantaneous rates (eg, substitutions of amino acids per site per evolutionary distance unit) that describes evolutionary change with a random or stochastic process. 15 Generally, these models invoke homogeneous Markov processes that assume that the probability of character state change does not depend on the probabilities of change of other characters, the previous history of that character, or the branches (timeframe) of the phylogeny in which change occurs. Many of these assumptions are unlikely for some characters, especially those that describe features that interact with each other to form higher levels of biological organization. 16 For example, amino acids interact to form secondary, supersecondary, and fold structure in proteins, and their change is constrained not only by these interactions but also by the role they play in imparting function. Their role is also likely to have changed in evolution as molecular interactions were being established. Thus, a Markovian model may not portray constraints imposed by the history of interactions. Furthermore, evolution destroys information through the impact of branching and the Markov chain convergence, especially under high mutation rates typical of sequences.17,18 Thus, sequence analysis is only useful for studying relatively recent divergences and its effectiveness decreases as we go deeper in time.

Typical character state transformation models used in phylogenetic analysis. (A) Character state graphs (CSGs) for equally weighted undirected characters with four character states (0, 1, 2, and 3). Edges describe allowed transformation between character states. The CSG in the left is a typical maximally connected character, an “unordered” character, while the CSG in the right is a minimally connected character embodied in a “fully ordered” character. The CSG in the middle is a partially ordered CSG containing a reticulation. (B) Transformation between character states can be undirected or directed depending on the costs D ij applied to the transformation from character state i to state j, or vice versa, with i ≠ j. (C) Character state matrices (step matrices). The matrices show state indices describing transformation costs (in tree lengths) from one character state to another. The Wagner and Fitch models use static ordered (additive) and unordered (nonadditive) characters, respectively. The Camin-Sokal, Dollo, and Arbitrary models involve asymmetric stepmatrices with transformation costs that violate the triangle inequality, a necessary property of phylogenetic distances. In Camin-Sokal optimization, reversals are prohibited by taxing them with an infinite cost. In the Dollo model, N is such that each gain of a character occurs only once on a phylogeny. In the arbitrary model example borrowed from Harish et al, 13 gains are taxed more than loses with an idiosyncratic asymmetric step matrix.
Second, the phylogenetic implementation of axiom 2 implies the construction of phylogenies. A phylogeny is a historical statement, preferably unambiguous, that generally takes the form of a tree or network, depending on the absence or presence of reticulations, respectively (Figure 2). It considers a multidimensional relationship of characters and taxa. This relationship defines a historical succession of singularities of character transformations leading to extant or extinct taxa. A phylogeny must also have a cost derived from some objective function, which serves to evaluate its quality. In the absence of reticulation, the branches of the network make a tree structure that unfolds from the root to its leaves (terminal nodes) as evolution proceeds. 15 Note that a phylogenetic tree must be rooted to fully explain the evolutionary process and that rooting implies a single evolutionary origin of taxa and a series of symmetry-breaking (cladogenetic, speciation, or furcation) events. Each internal node of the tree represents an ancestor to sets of other more derived ancestors and taxa (these sets are known as “clades”). In the presence of reticulations, single or multiple origins can lead to a network structure that finally resolves into leaves.19-22 A network of this kind implies the existence of explicit convergent and divergent relationships in the historical statements, with changes occurring simultaneously in some regions of the network (“parallelisms” or “convergences”). It also implies the existence of symmetry-joining events.

Phylogenetic trees and networks. (a) An unrooted tree and a corresponding network describing the phylogenetic relationship of four taxa (labeled A, B, C, and D). Note the reticulation connecting terminal branches leading to A and B through internal nodes of the network. The arrow indicates one of many branches that can be pulled down to root the tree and network. (b) Rooted tree and corresponding network. The branch that was pulled down during rooting now contains the root node and has polarized character changes in the tree and network structure by defining an “arrow of time.” Internal nodes are now ancestors of nodes that are derived from them. The reticulation in the rooted network implies the existence of character changes occurring in parallel.
It is noteworthy that a tree representation is a coarse-grained historical account of a network. The tree hides any evolutionary processes of reticulation that have coexisted with a vertical pattern of descent with modification. This coarse-graining property can simplify the burden of computing character change in alternative network topologies. Our focus from now on will be trees rather than networks for methodological, computational, and other reasons. Trees can be constructed most effectively using search methods (eg, tree bisection and reconnection) that explore the multiple sub-spaces of all possible tree topologies and select the local maxima according to some optimality criterion. 15 The optimality criterion in maximum parsimony selects trees that entail the least amount of character state change, with one or many trees being optimal. One widely adopted procedure minimizes the Manhattan metric that measures distance in abstract multidimensional spaces, with distance corresponding to number of independent origins of characters. Maximum likelihood methods search for trees that are most likely to occur, given the probabilistic evolutionary model, while converging to a single hypothesis (tree). Bayesian methods select a range of trees according to their posterior probabilities, given data, model, and prior probabilities (belief) on the historical hypotheses, instead of searching for the optimal tree. The selected trees are used to reconstruct a consensus tree, where support strength of individual clades is represented by the posterior probability. 23 We note that finding the best estimate of phylogeny is a non-deterministic polynomial-time (NP)-hardness problem. For example, exhaustive or branch-and-bound algorithmic implementations allow exact maximum parsimony solutions when the number of taxa is less than 20. However, the dimensionality of the space of unrooted (u) and rooted (r) trees increases with number of taxa n according to N u = (2n − 5)!/2n − 3(n − 3)! and N r = (2n − 3)!/2n − 2(n − 2)!, respectively. A space of rooted trees with only 50 taxa contains 2.7 × 1076 possible trees, which exceeds Eddington number of electrons in the visible universe. Thus, building large phylogenies is computationally demanding. It requires heuristic searches, often with hill-climbing algorithms of tree space exploration, including ratchet, genetic, and divide-and-conquer algorithms and simulated annealing. 24
Third, phylogenetic implementation of axiom 3 implies the identification of characters in evidence. The way how characters are shared between taxa implies an “homology” relationship between character states. Given a phylogeny, this relationship impacts the phylogenetic signal that can be extracted from evidence. Owen initial definition was structural and independent of history, ie, the simple appearance of a feature in different taxa implied a homology. As the functional and historical aspects of this “sameness of structure” criterion were not considered, a same structural feature could have had separate origins. The feature could have converged to the same structure in evolution or could have resulted in unrelated functions. Thus, a historical and ontological definition of homology was needed. This was provided by Osborn and made analytically explicit by Hennig with his concept of “shared and derived” character relationships (“synapomorphies”). Thus, homology is currently and appropriately equated to common ancestry, descent from a common ancestor, or even better “a unique origin for each derived condition,” 1 when origins are interpreted broadly to include loss. If all characters would be of this kind, there would be no conflict and a “true” phylogeny would logically follow. However, not all characters are phylogenetically useful and some are relatively more prone to lose informative signal over time. 17 Some represent true homologies, while others embody “homoplasies,” similarities that do not result from common ancestry but arise from multiple origins. In fact, congruent sets of characters are hardly free of homoplasy, and there is no data type that leads to a true phylogeny. The implication of this fact is both conceptually and operationally fundamental. 1 It was already made explicit by Wiley. 8 Homoplasy results from deficits of the phylogeny in its ability to convey the true historical ground plan. This may stem from our ignorance of its complexity or the evolutionary processes responsible for it. Homoplasy may also result from incorrect definitions of characters and model or simply because characters in evidence contain weak or frustrated phylogenetic signal. Thus, postulating ad hoc hypotheses of homoplasy disposes of evidence against a phylogeny and its supporting synapomorphies. Consequently, the operational implementation of phylogenetic parsimony minimizes the number of logically independent ad hoc hypotheses of homoplasy. 1 This central tenet of phylogenetic analysis aligns with the hypothetico-deductive framework of scientific inquiry, which seeks avoidance of ad hoc assumptions in hypothesis testing.
The Consequences of Phylogeny Reconstruction
The consequences of the three evolutionary axioms that we have described are conceptually and operationally interlinked. Establishing homology requires a phylogeny and a criterion of character polarization. Selecting a phylogeny demands minimizing ad hoc hypotheses of homoplasy in the ensemble of all possible phylogenies. It also requires establishing an appropriate transformation series or model. Selecting useful characters implies assuming they represent homologies and later confirming their homology relationship within the congruent character set. This requires unfolding transformational change in the branching patterns of the phylogenetic trees, which must be rooted by establishing an “arrow of time.” Rooting implies identifying the plesiomorphic and apomorphic transformational homologs. These multiple interrelationships make the entire retrodiction enterprise an endeavor of reciprocal fulfillment. In this process, two auxiliary principles have been enormously helpful. Both establish that nothing must be prohibited a priori unless there is evidence to the contrary:
Hennig Auxiliary Principle. This principle prompts always assuming homology in the absence of contradicting evidence. 2 It provides a “discovery mechanism” to identify putative homologies by induction using the world of experience of the investigator. 8 Similarities and dissimilarities are first identified, using, eg, ontogenetic or positional correspondence relationships or even machine-learning techniques of classification. Trivial hypotheses are first excluded, but those that remain are then put through phylogenetic test. The goal is to increase explanatory power and validity of individual phylogenetic hypotheses. This process of “reciprocal illumination” 2 between each “primary homology” statement and favored phylogenies obtained from all available data results in “secondary homologies,” homologies that have been put to the falsification test and have proven their mettle. 25 This scheme for developing scientific theories of evolution adds additional evidence in the form of more informative phylogenetic characters and taxa to a corpus of growing ideographic evidence.
Kluge Auxiliary Principle. A second and equally useful criterion is the principle of always assuming character independence in the absence of contradicting evidence (following Brooks and McLennan 26 ). Characters should reflect independent pieces of historical evidence:27,28
“If two characters were logically or functionally related so that homoplasy in one would imply homoplasy in the other, then homoplasy in both would be implied by a single ad hoc hypothesis. The “other” homoplasy does not require a further hypothesis, as it is subsumed by the relationship between the characters. This is the principle underlying such common observations as that only independent lines of evidence should be used in evaluating genealogies …”
1
Thus, any co-variation induced by interactions between the characters that are being studied (structural, physiological, developmental, behavioral, etc) complicates the phylogenetic reconstruction. Dependencies can also result from logical correlations arising from the definition of characters, including their ontology. Character interaction in evolution results in characters being overweighed in the analysis. 29 These dependencies distort and obscure phylogenetic signal. They must be either encoded into the phylogenetic model through parameters or weight corrections or avoided by excluding at least one of the offending characters from the data matrix. As phylogeny fails to represent true history when dependencies are not made explicit or avoided, it is important that they be appropriately evaluated. Kluge principle protects phylogenetic analysis from a priori maneuvers of character weighting or character exclusion in the absence of knowledge about character non-independence. This principle provides a starting point of phylogenetic discovery in light of the existence of cohesive networks of interactions that establish at different levels of the hierarchy of biological organization. 30
Minimizing Ad Hoc and Auxiliary Hypotheses
The hypothetico-deductive scientific method demands that competing hypotheses be judged on the basis of observation. Ad hoc and auxiliary hypotheses are assumptions, sometimes unwarranted, that dispose of conflicting observations. For that reason alone, the practice of minimizing them is widely accepted in the empirical sciences. For Popper, 31 an hypothesis is ad hoc if it is introduced with the sole purpose of explaining “a particular difficulty that cannot be tested independently.” In contrast, an auxiliary hypothesis has the same purpose, but its conjecture can be tested independently from the main hypothesis. In other words, it represents a hypothesis other than the test hypothesis which is assumed to be true and is needed to derive the test implication. The existence of ad hoc and auxiliary assumptions is philosophically important for confirmation of theories. When overthrowing theories, the problem is to distinguish the disconfirmation of a hypothesis from that of its auxiliaries. This well-known problem of falsificationism is embodied in Duhem-Quine thesis, which posits that it is impossible to test hypotheses in isolation and that the enterprise is really a joint test of the hypothesis and its associated assumptions. 32 The criticism has been recently lifted by arguing that auxiliary hypotheses can be themselves subject to measures of corroboration, a maneuver that amounts to establishing priors in the Bayesian confirmation of theories.33,34 By the same token, the adoption of ad hoc auxiliaries is accompanied by hypothesis disconfirmation when the “improbable antecedently” auxiliaries are conjoined with the hypothesis. 35 This allows to recast in Bayesian terms concerns about unfalsifiability as they relate to formal and argumentative ad hocness. Formal ad hocness has a negative effect on a theory, but its disconfirmation “cost” can be calculated. Argumentative ad hocness has in addition some degree of irrationality or failure of reason that does not provide an explicit cost. This distinction is philosophically important not only for lifting the Duhem-Quine thesis but also when considering auxiliaries in the presence of apriorism, ie, the practice of salvaging belief by making hypotheses unfalsifiable.
In phylogenetic analysis, ad hoc hypotheses of homoplasy are minimized when searching the space of possible trees and selecting for competing hypotheses of history. Specifically, these auxiliaries represent hypotheses of multiple origins that are not accounted for by the tree representation of history, the evolutionary model, and their match to character evidence. Their existence rescues our inability to appropriately model evolution. As the number of homoplasies serves as optimality criterion for choosing the best phylogenetic tree, their disconfirmation cost is explicitly calculated (eg, with metrics such as the consistency index [CI]). They embody formal ad hocness. When calculating this cost, the independence of ad hoc hypotheses of homoplasy discussed above is of particular concern. Similarly concerning are homologies that fail corroboration.
Auxiliary assumptions are also important for phylogenetic analysis. They not only affect homology and homoplasy determinations, ie, the relationship between history and ad hocness, but also the ability to overthrow historical hypotheses, ie, their falsifiability. Within the hypothetico-deductive method, hypotheses must be subjected to “severe” tests.
36
The logical relationship between a hypothesis of history (a tree or network) and the evidence in the form of putative homologies makes only sense in relation to what is known, background knowledge. The logical improbability of the test hypothesis defines its potential to be tested, its testability. In other words, corroboration of a hypothesis by evidence requires that that evidence be improbable given background knowledge alone. The demand than the hypothesis be improbable on background knowledge can be illustrated with Kluge example of a three-taxa historical statement, with taxa labeled A, B, and C: “Given only descent with modification as the background knowledge, synapomorphies characteristic of (A,B), (A,C) and (B,C) should be equally likely… However, if a large majority of one class of those possible synapomorphies were to be discovered, say that which characterizes hypothesis (A,B), then this is unlikely given the background knowledge alone, but not under the background knowledge plus the postulated rooted (A,B)C cladogram. The (A,B)C hypothesis is said to be corroborated to the degree to which those (A,B) synapomorphies are observed.”
36
Increasing the number of characters will increase the number of independent tests and therefore severity of test. Similarly, increasing the number of taxa will increase the universality of the historical statements, the chances of disconfirming evidence, and the “boldness” of the historical hypothesis. In contrast, severity of test decreases when background knowledge is increased by invoking auxiliaries such as adding an assumption of rooting (see below), a priori weighting of characters or character transformation costs compatible with some form of phylogenetic congruence, or considering assumptions of pattern and process. Furthermore, in the presence of competing hypotheses, the number of shared auxiliary assumptions should be maximized, thereby enhancing the attribution of evidence to the main hypothesis. The course of action of adding auxiliaries, however, should be avoided at any cost. “Adding to background knowledge is a verificationist slippery slope, which ultimately ends in tautology.” 36 Thus, unproblematic auxiliary assumptions in background knowledge must be minimized. Such minimization (1) increases severity of test, (2) attenuates the Duhem-Quine problem, and (3) increases simplicity and boldness. “Bold ideas, unjustified anticipations, and speculative thought, are our only means for interpreting nature: our only organon, our only instrument, for grasping her.” 31
Rooting Criteria in Phylogeny Reconstruction
A phylogeny must be rooted to portray history. This involves orienting an unrooted tree by identifying and “pulling down” a branch (edge) that will hold the ancestor of all taxa examined. However, rooting brings with it ad hoc and auxiliary assumptions, which could weaken retrodiction. Kluge three-taxa statement example discussed above highlights the importance of rooting in phylogenetic analysis. Fundamentally, the rooting of trees is necessary to unfold the full frustrated interplay of homology and homoplasy, evaluate tree building optimality of rooted trees, and build character state vectors of ancestors. As mentioned earlier, characters that unfold in phylogenies as homologies must show at least two transformational homologs, one ancestral and another derived. In cases where these “dynamic” homologies involve multistate taxa, polarization has the potential to reveal nested patterns of the multiple transformational homologs. Polarization of character state changes, whether performed a priori, a posteriori, or during tree reconstruction, roots the recovered optimal phylogenies. This is a necessary and sufficient property of phylogenetic inference, which unfortunately has been neglected in many phylogenetic and phylogenomic studies. Several rooting approaches are available that make use of formal auxiliary hypotheses. These approaches have been classified into two main groups by Nelson: 37 indirect and direct methods. Indirect methods require character information from taxa external to the study group (the ingroup). In turn, direct methods focus exclusively on ingroup taxa. Indirect and direct methods also differ in that inferences of character states are made at two different nodes 38 (Figure 3). Indirect methods focus on the outgroup node. This node is separated from the ingroup clade by one internode and represents the most recent common ancestor of the ingroup and its most closely related outgroup. In turn, direct methods focus only on the ingroup node, the most recent common ancestor of the ingroup. Argumentative ad hoc and auxiliary hypotheses have been also used to root trees using indirect and direct methods in numerous studies. As we will explain in the following, these approaches should be avoided because they undermine the testability of phylogenies and because they fuel apriorism in ideographic analysis. Table 2 summarizes rooting strategies we will now describe.

Rooting trees and polarizing character state transformations. In outgroup comparison, the occurrence of character state X is diagnostic of the outgroup and is used to root the tree by assuming the root is not located in the ingroup. Once the outgroup is made ancestral, the tree is rooted and character state Y is shared and derived, making it a synapomorphy. In Nelson ontogenetic criterion, the character state distributions in two ontogenetic stages are used to polarize character transformation. Character state X is more generally distributed than Y within the ingroup because X is present in all taxa and Y is present in only a subset. Thus, character state X is ancestral to Y, and Y is a synapomorphy. To satisfy Nelson rule, ontogenetic stage 0 must precede stage 1. In Weston generality criterion, Nelson rule is extended to any case, including the ontogenetic and paleontological method. Character state Y is less distributed than X and is considered derived. Character state Y is also shared and derived. In the paleontological method, the earliest known fossils of the ingroup have character state X and are used to root the tree. The figure was modified from Bryant. 39
Rooting strategies.
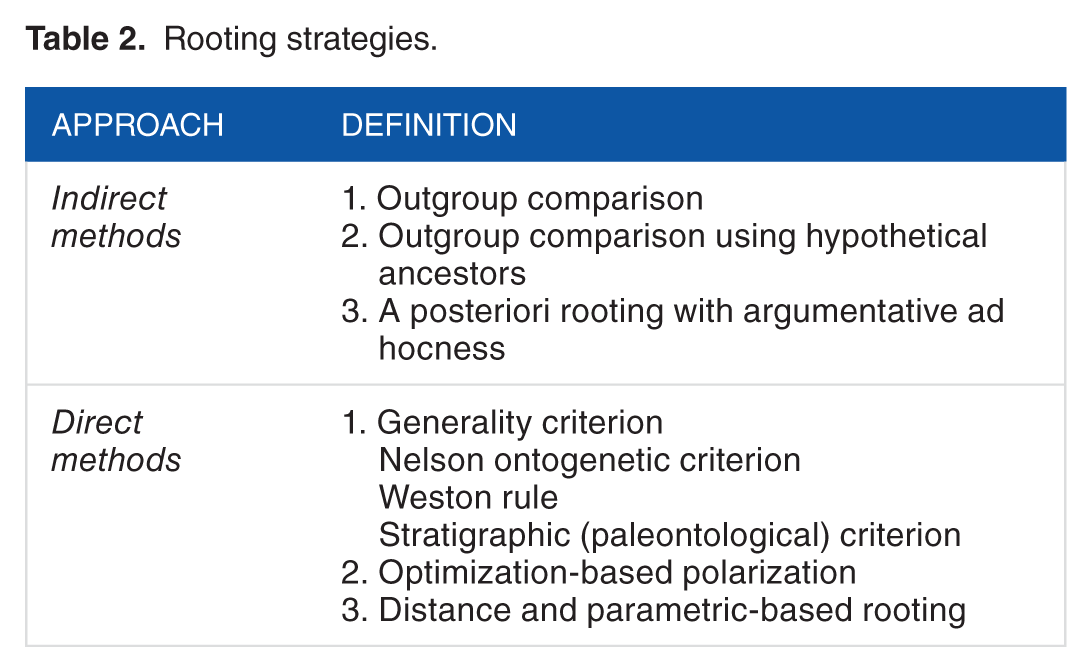
Indirect methods
Indirect methods generate an unrooted tree by optimization and then select a taxon subset, which is defined a priori as being of more ancestral origin. This subset is pulled down to the base of the tree. At least three auxiliary assumptions support indirect methods: (1) higher level relationships are outside the ingroup, (2) equivalent ontogenetic stages pertaining to the developmental history of an organism are compared, and (3) character state distributions are appropriately surveyed. 39 All methods root trees a priori by either selecting an outgroup with a proper character state distribution that is more inclusive or defining a hypothetical ancestor, which is then used as outgroup to create an outgroup node.
Outgroup comparison
In outgroup comparison, rooting is inferred by the distribution of character states in the ingroup and a sister group, which includes a taxon or set of taxa external to the ingroup.38,40,41 This outlying group is known as the “outgroup.” In the most simple case, if the character state is only found in the ingroup, the state is considered derived and character state distributions provide a basis for rooting the tree. Currently, trees are rooted with outgroups after building unrooted trees with search methods that include ingroup and outgroup taxa. These methods identify the edge that leads to the outgroup that is closest to the ingroup and create a outgroup node (a new vertex) for orienting (rooting) the phylogeny. Character distributions in multiple outgroups can be summarized in the outgroup node (Figure 3). While outgroup comparison is by far the preferred method because phylogeneticists tend to have confidence in the supporting assumptions, the method can be problematic.
Outgroup addition is usually a priori and ad hoc. It adds a minimum of an additional character state vector that is assumed to be ancestral, which epistemologically can add uncertainty about the relation of the outgroup to the ingroup (see Lundberg 42 ). The inductive reliance of outgroup addition on assumptions of higher level relationships can lead to infinite regress or apriorism (especially in ToL reconstruction). The increase in the level of universality of the phylogenetic statements has also consequences during optimization of phylogeny reconstruction. For example, additional character state vectors can increase ad hoc hypotheses of homoplasy that could affect ingroup relationships (especially if the outgroup are taxa that are evolutionarily distant from the ingroup; eg, Graham et al 43 ). Unless well justified, outgroups can be not only problematic but impossible. They cannot root the ToL or groups of organisms isolated by organismal diversity that has not been appropriately surveyed (biodiversity “dark matter”). 44 Finally, the outgroup comparison method in itself does not polarize characters and root trees. It simply extends the tree by connecting the ingroup to the rest of the phylogeny.45,46 The tree is rooted by reasonably assuming that the root is not located within the ingroup. Despite these shortcomings, outgroup comparison is the method of choice in phylogenetic analysis and has helped enormously in the efforts of systematic biology.
Outgroup comparison using hypothetical ancestors
A hypothetical ancestor can be used as outgroup. The ancestor can summarize in its character state vector the character state distribution of outgroup taxa or can represent an artificial taxon selected based on other assumptions. The practice can add auxiliary assumptions of unproblematic background knowledge, other than those previously specified, 39 argumentative auxiliaries, and/or additional ad hoc hypotheses of homoplasy during phylogenetic optimization. While the use of these hypothetical ancestors can be justified, Bryant 47 cautions that the hypothetical ancestor should not combine inferences based on outgroup comparison with those based on generality, ontogenetic, paleontological, and other direct methods. Inferences regarding plesiomorphic states apply to the outgroup or ingroup nodes and should be combined into a single hypothetical construct. Hypothetical ancestors are usually treated as taxa. The implication of their use is that they represent extant or extinct biological entities, not ancestors per se. Some studies have used hypothetical “all-zero” pseudo-outgroups as a strategy to root trees. However, the assumption is a priori and can be risky if not adequately supported. An interesting refutation is illustrated by Wheeler, 15 which shows the misplacement of Heteroptera insects in different families when using this strategy.48,49
A posteriori rooting with argumentative auxiliary hypotheses
Outgroups and ancestors have been treated as argumentative auxiliary hypotheses to identify a branch of an unrooted tree, which is either annotated or pulled down to root the phylogeny. This is done without seeking the benefits of phylogenetic optimization of any kind. The approach should be avoided because it represents a notorious apriorism (eg, Williams et al 50 ).
Direct methods
Direct methods seek phylogenetic optimization of character state information pertaining to ingroup taxa. They can be supported by the single auxiliary hypothesis that character state distributions in the ingroup are appropriately surveyed. 39 Some methods root trees a posteriori by first reconstructing an unrooted tree from additive (ordered or continuous) or nonadditive (unordered) static characters (Figure 1) and then polarizing them with implementations of the Lundberg method 42 (see in the following). Other methods polarize characters directly during optimization with a character state matrix of arbitrary transformation costs, which brings additional auxiliary hypotheses and requires dynamic programming during phylogenetic reconstruction. Still other defines ancestral states a priori, such as those that make use of the fossil record.
The generality criterion
The generality criterion embodies Nelson ontogenetic criterion, Weston methodological rule, and the stratigraphic method. The ontogenetic criterion and Weston rule are the most powerful rooting methods available. They are based on the distribution of homologous character states in the nested hierarchy of ingroup taxa and use the minimum number of unproblematic auxiliary assumptions.
Nelson 37 ontogenetic criterion is a special case of the generality criterion. It is restricted to morphological characters expressing in ontogenetic stages but the method could be extended to molecular markers of development. The method was inspired by the assumption that ancestral character states occur earlier in ontogeny than derived states and that character state changes occur by “terminal addition.” 2 In other words, the distribution of the states of homologous characters in ontogenies of the ingroup confers polarity through the generality of character states, with more widely distributed states being ancestral (Figure 3). Nelson rule (his biogenetic law) makes this explicit: “given an ontogenetic character transformation from a character (state) observed to be more general to a character (state) observed to be less general, the more general character (state) is primitive and the less general advanced.” 37 An illustrative example is the vertebrate endoskeleton of the shark and perch, which is cartilaginous in the early embryos of both species and then differentiates into bone only in the perch. Thus, cartilage is the ancestral character state. Unfortunately, Nelson “generality” has been the subject of multiple interpretations and non-productive debate.39,51 The “hierarchical” concept of generality has been confused with the “frequency” concept of commonality. It has been interpreted as a sequence of ontogenetic change when in fact it is an expression of ontogenetic character transformations between alternative character states in different ontogenetic stages, with the one that is more widely distributed considered ancestral (Figure 2). The method was originally conceptualized for vertebrate phylogeny. It involves comparison of developmentally nested and distinct life history stages, which generally cannot be implemented, eg, in the study of microbial organisms.
Weston45,52 realized that the ontogenetic criterion embodied a wider and more universal generality criterion in which the taxic distribution of a character state was a subset of the distribution of another. He realized that as long as ancestral character states were preponderantly retained in descendants, they will always be more general than their derivatives given their nested hierarchical distribution in rooted phylogenies. In other words, character states that characterize an entire group had to be considered ancestral relative to an alternative state that characterizes a subset of the group (Figure 3). Weston more general rule therefore specifies the following: “given a distribution of two homologous character (states) in which one, X, is possessed by all of the species that possess its homolog, character Y, and by at least one other species that does not, then Y may be postulated to be apomorphous relative to X.” 52
The generality criterion is based on the fact that every homology is a synapomorphy (shared and derived feature) in nature’s nested taxonomic hierarchy and that homologies in the hierarchy result from additive phylogenetic change. 45 We interpret additive change as the successive origination of new character states by innovation and their spread in an unfolding phylogeny. The generality criterion can be readily visualized when applied to cases in which homologous entities accumulate “iteratively” in evolution. In the example of Figure 4, a multistate character (a serial homology) adds character states in time and in doing so spreads differentially in a tree. The evolution of states of parts (characters) manifests in the evolution of wholes (taxa). Note, however, that serial homologies can be decomposed into their component homology parts and that the nesting patterns will be maintained. This represents a fundamental property of evolution.

The basics of the generality criterion. In evolution, character states originate in time by terminal addition. This implies a comb-like rooted tree in which basal splits describe the origin of states that are ancestral and splits closer to the crown describe the origin of those that are more derived. A tree (left) portrays the evolution of a three-state multistate character, with states labeled 1, 2, and 3. When this character is traced onto a tree describing the evolution of a set of four taxa (labeled A, B, C, and D; right), character state evolution manifests in the nested lineages. The tree shows an example of how the states of the three-state character unfold in its internal and terminal nodes. Ancestral states are more popular than derived states as character evolution must unfold within the nested branches. This generality can be used to root phylogenetic trees. Note that any multistate character can be decomposed into two-state character components, which will continue to comply with the generality criterion. Also note that the origin of character states is independent of the transformational dynamic of the homologs. For example, characters’ transformation can be made fully reversible and the general nesting patterns will maintain.
The iterative accumulation of homologies implies increases in biological abundance. This process of accumulation and retention of iterative homologs occurs at different levels of biological organization and is a conclusion, not a premise of Weston rule. For example, in serial homology, existing biological structures are gradually modified by discrete intermediary steps. For example, body segments of animals, such as the development of forelimbs and hind limbs of tetrapods or the iterative structure of the vertebrae, are the result of the duplication of master control regulatory genes such as homeobox-like genes followed by their divergence. This results in major stepwise morphological evolutionary transformation. For example, Gegenbaur classical hypothesis of jaw-gill arch serial homology has been recently confirmed and linked to the nested expression of Dlx genes in vertebrates. 53 Thus, higher level structures unfold iteratively by the recurrent action of lower level molecular structures. An example of serial homologs in molecular biology is paralogous genes, genes that spread in genomes by duplication and divergence. Paralogous genes have been used effectively to root trees, beginning with Schwartz and Dayhoff. 54 As paralogy and orthology cannot be resolved without phylogenetic analysis, the sequences of putative orthologs and paralogs in a set of taxa are aligned and analyzed. Remarkably, the analysis simultaneously resolves orthology from paralogy and also roots the subtree for each set of orthologous sequences. Thus, one paralog acts as an outgroup of the other when both are included in the phylogenetic reconstruction. We note that paralogy is equivalent to serial or mass homology in morphology. However, multiplications occur in phylogeny in the former and in ontogeny in the latter.
The stratigraphic (paleontological) criterion of geological character precedence establishes that characters states of older fossils are ancestral when compared with those of younger counterparts. Thus, the oldest known fossil taxon in the ingroup directly roots the tree. Similarly, the tree can be rooted with a hypothetical ancestor that summarizes the character state vector of fossil taxa of the ingroup. The stratigraphic method is problematic. It relies on a number of auxiliary assumptions when fossils are available, including the completeness of the fossil record, that fossil evidence belongs to the ingroup, that fossil age assignments are correct, that equivalent ontogenetic stages are being compared, and that character state distributions in the ingroup are appropriately surveyed. 39 However, the stratigraphic rationale can still be powerful in establishing molecular links between evolution and development (eg, Domazet-Lošo and Tautz 55 ), gene generation (eg, Carvunis et al 56 ) and dating of the ToL. 57
Operationally, the generality criterion can be satisfied by reconstructing optimal unrooted trees for ingroup taxa and rooting them a posteriori using the Lundberg rooting method. 42 This method finds the internode at which a hypothetical ancestor can be attached most parsimoniously. The hypothetical ancestor provides the directionality needed for terminal addition of Nelson or Weston rules or summarizes the criterion of geological character precedence. Optimization during rooting complies with the optimality criterium used for tree reconstruction. In all cases, Lundberg rooting considers only character state distributions within the ingroup, pulls down the ingroup internode most parsimoniously, and polarizes character state change. Thus, Lundberg 42 differentiates the direct and indirect methods by focusing on ingroup taxa and optimizing character state vectors of the ingroup node with a hypothetical ancestor that is not included in ingroup tree optimization. This maneuver links unrooted tree optimization of modern phylogenetic analysis and Hennigian recognition of archetypal ancestors within the framework of the generality criterion.
Optimization-based polarization
The assignment of character state vectors to internal nodes (optimization) in maximum parsimony reconstructions does not rely on a stochastic model of character state change. Instead, it requires a transformation cost matrix (step matrix) that specifies the costs (distances) of all possible transformations between character states. Static character types such as additive (also known as ordered or Wagner), nonadditive (unordered or Fitch), and matrix characters (general or asymmetric) are computationally optimized in polynomial time from taxon-fixed character state vectors (Figure 1). However, distances are minimized/maximized during optimization over all transformation elements. Bounded optimization requires that distances be “metric” by satisfying four specific mathematical conditions (Table 3). Distances can only faithfully represent trees if they are additive and satisfy an additional four-point condition. Finally, some additive distances are also ultrametric and result in rooted trees, which exhibit a “molecular clock” property along their branches. Asymmetric stepmatrices (eg, Camin-Sokal, Dollo and arbitrary; Figure 1) also produce rooted trees. However, their distances are not metric. They fail the triangle inequality condition, which impacts the validity of phylogenetic reconstruction (eg, Wheeler 58 ). They also require justification. Every arbitrary transformation cost embodies auxiliary hypotheses joining the test of a historical hypothesis, weakening its falsifiability.
Conditions of metric, additive, and ultrametric distances (costs) used in tree optimization.

Distance and parametric-based rooting methods
Midpoint rooting calculates all leaf-to-leaf distances and places the root half-way between the most distantly separated leaves. 59 The method relies on the assumption of a reasonable “clock-like” rate of evolution across all branches of the tree. It works best with a well-balanced tree but is highly susceptible to unbalanced rate heterogeneities. It can provide misleading results if the root is placed within a dense set of short branches. Interestingly, an empirical comparison of outgroup and midpoint rooting suggests a correlation between their consistency in selecting a root. 60 While distance methods that measure overall similarity or dissimilarity can create rooted trees, they do not exhibit the desirable properties of character-based methods. They cannot reconstruct character state vectors at internal nodes and changes in edges and cannot establish ancestral-derived relationships that would test statements of homology and the rooting hypotheses. In contrast, character-based methods derive internal node vectors and spanning branch distances (maximum parsimony), probabilities of edge transformations and time parameters (maximum likelihood), and integrations of the distributions of model and time parameters (Bayesian methods). Parametric methods can root trees by assuming a molecular clock or by using a non-reversible substitution model. 61 Simulations showed the performance of strict or relaxed clock models can be superior to outgroup and midpoint rooting. 62 Rooting using the Bayesian framework with a Yule prior on tree topology (implemented in BEAST) is quite popular, 63 but there is a wide variety of methods that use relaxed clocks. 64 Many prune branches, divide global rates into local rates, and make trees partially ultrametric while correcting for rate heterogeneities. Others incorporate rate heterogeneities by estimating branch length without assuming rate constancy and then apply modeling strategies that minimize length discrepancies over the branches.
Farris 65 objected to the molecular clock idea with the simple and powerful argument that if a clock existed, distances would be ultrametric. However, non-trivial ultrametric data are inexistent or most rare. Furthermore, transforming real distances (often additive) with stochastic models to offset the effects of saturation or back mutations in sequences and account for total change results in loss of metricity (violating the distinctness and triangle inequality conditions). 15 In the absence of ultrametricity, the use of a relaxed clock could still salvage the rooting strategy for some data if the prior probability of the topology of the tree would carry the location of the root and an optimal unrooted tree is known. 66 These theoretical arguments must be, however, validated with empirical studies.
Methods that reconcile the space of gene trees with alternative rooted species trees under a joint probabilistic model of sequence evolution, such as the amalgamated likelihood estimation (ALE) method, 67 enable to root species trees with the help of gene duplications, transfer, and loss events. 68 However, the robustness of these methodologies remains to be properly evaluated in the presence of varying levels of gene events, “small genome attraction” artifacts that favor roots that divide smaller from larger genomes on the tree, and variations in the probabilistic models.
Rooting the ToL
The reconstruction of a ToL depicting true organismal biodiversity is hampered by both the enormous scope of the problem and the challenges of phylogenetic analysis. While there are more than ~1.8 million named species (eg, Mora et al 69 ), it is estimated that there are more than one trillion (1012) microbial species on Earth. 70 In addition, unknown levels of biological “dark matter” exist that have not been surveyed. 44 Only recently, uncultivated and little known organisms have been added onto expanded ToL constructions. 71 Integration of thousands of published phylogenies summarizing evolution of ~2.3 million taxa and more than ~0.2 million internal nodes (while preserving conflict) still provide patchy evolutionary views with poor resolution (with an average of 16 children per node). 7 Important conflicts exist, including the contentious monophyly of Archaea and its relationship to Eukarya, multiple resolutions of early diverging eukaryotic and animal taxa, hyperdiverse and poorly understood organismal groups (Archaea, Bacteria, basal eukaryotes, and fungi), and the place of viruses in the ToL. The use of outgroups to root subtrees is limited by notorious problems in identifying the root of major organismal groups including birds, mammals, and angiosperms.72-74 In addition, the problems of holobionts and the species concept,75,76 especially in akaryotic groups 77 prone to rampant horizontal transfer, 78 compromises the integrity of definition of taxa. Even our ability to dissect superkingdoms of life is limited by evolutionary understanding of levels of biological organization. For example, evolutionary statements for the origin of eukaryotes derived from concatenated sequence analysis of highly conserved and presumably universal genes contradict those derived from embedded protein structural domains. 79 Most of the studies of gene sets or genomic repertoires have produced unrooted ToLs and should be considered incomplete statements of evolutionary history.
The rooting of the traditional (sequence-based) ToL remains contested despite of four decades of intense research following the discovery of Archaea. 80 While the outgroup method has established itself as the most common technique for rooting phylogenies, the absence of an adequate outgroup makes it impossible to root the ToL with these kinds of methods (see below). The first attempts to root the ToL employed paralogous gene sets that root each other and were believed to have diverged by gene duplication prior to the common ancestor of cellular life.81,82 Note that this methodology, which was first introduced by Schwartz and Dayhoff, 54 is a direct rooting method that complies with Weston rule. Initial paralogous gene sets were ATPases (α and β subunits) and elongation factors EF-Tu and EF-G. The method was quickly extended to a number of additional paralogous gene couples (reviewed in Zhaxybayeva et al 83 ), including elongation factors EF-1 and EF2, aminoacyl-transfer ribonucleic acid (tRNA) synthetases, signal recognition particle proteins, aspartate and ornithine transcarbamoylases, carbamoyl phosphate synthetases, and histidine biosynthesis genes. A similar paralog-based top-down rooting approach considers both insertion-deletions (indel) and gene gains and losses in incomplete gene sets (reviewed by Lake et al 84 ). The methodology was introduced with the well-studied indel of Hsp70/MreB gene sequences. 85 While many of these studies favored a rooting between Archaea and Bacteria, gene sets rooted ToLs differently, including several root positions within Bacteria. The methodology has been shown to be severely compromised by a number of problems and artifacts of sequence analysis, including long branch attraction, mutational saturation, taxon sampling bias, horizontal gene transfer, and hidden paralogy.86,87 More troubling is the problem of historical segmental heterogeneity of gene sequences,44,88 which affects the validity of the use of gene sequence alignments in general (including concatenated sets) in evolutionary studies. Alignments are built without recognizing the differential history of the modular structure of macromolecules, such as structural domains of proteins. Structural domains are the evolutionary and structural units of proteins, and their inception has been occurring since the origin of proteins. 89 The existence of domains is neither considered in sequence alignments nor considered in evolutionary models for alignment and phylogeny reconstruction. We illustrate the problem with a subset of a famed concatenated sequence alignment of universal molecular sets that was used to support a two-superkingdom model of diversification of life. 90 The alignment included elongation factors, aminoacyl-tRNA synthetases, ribosomal proteins, and ATPases. A simple mapping of domains’ structural cores defined by advanced hidden Markov model (HMM) libraries of structural recognition onto the sequence alignment of its concatenated genes shows irreconcilable misalignments that compromise the integrity of structural domains and challenge the validity of the alignment exercise (and its associated tree reconstruction). Figure 5 shows a simple analysis of phenylalanyl-tRNA synthetase, one of the conserved sequences of the concatenated alignment. It reveals artifactual excisions of important regions of the enzyme molecules, such as the crucial anticodon-binding domain necessary for genetic code specificity. Despite bettering the alignment, even the exercise of trimming positions with >50% gaps (partial deletion) introduces serious uncertainties, especially because the exercise is highly dependent on the taxa included in the study. For example, Spang et al 90 included 84 Archaea, 10 Bacteria, and 10 Eukarya in their analysis. Hence, using a threshold of eliminating sites with >50% gaps is highly dependent on the presence of those gaps in Archaea. Ideally, taxa should be sampled from all groups to avoid such downstream ambiguities. This adds to the problem of taxa and character sampling of any phylogenetic analysis. Trimming can also potentially eliminate variable but central segments of structure that could carry significant evolutionary history. In this regard, a recent study revealed that contentious phylogenomic relationships at deep evolutionary level can be driven by a handful of sites in a handful of conserved genes of the concatenated sequence alignment. 91 Therefore, incongruent phylogenetic relationships must be carefully evaluated.

Structural domains challenge the validity of phylogenies derived from concatenated sequence alignments. (A) The HMM-driven assignment of domain families to phenylalanyl-tRNA synthetase (PheRS) arCOG00412 sequences of the concatenated alignment of Spang et al. 90 Multiple heterogeneities in protein length and domain makeup are visible, including variant domains at the N-terminal and C-terminal regions in Bacteria. We found that a total of 15 of the 36 COGs present in the trimmed alignment showed misalignment and/or mismatches of domains of many universal genes that distort the integrity of domains and the validity of sequence-driven phylogenetic statements. We illustrate the problem with the first gene of concatenated set. Sequences sampled: (1) Desulfurococcus kamchatkensis; (2) uncultured Marine Group II euryarchaeote; (3) Lokiarchaeum; (4) Bacillus subtilis 168; (5) Rhodopirellula baltica SH1; (6) Thermotoga maritima MSB8; (7) Homo sapiens; (8) Trichomonas vaginalis; and (9) Tetrahymena thermophila PT. Domains are defined using SCOP concise classification strings (ccs). (B) MAFFT sequence alignment of arCOG00412 sequences (925 amino acids in length) shows domain read-through is affected by addition of gaps. The histogram below shows the percentage of gaps in each column of the alignment. (C) The MAFFT alignment trimmed to remove positions with >50% gaps (571 amino acids in length) removes 38% of original sites but eliminates both the Myf domain (b.40.4.4) and the anticodon-binding domain of PheRS (d.58.13.1) from the analysis of the molecules and shortens, splits, and distorts the other domain structures (eg, the B3/B4 domain of PheRS, PheT [b.153.1.1; green] necessary for tRNA binding was eliminated in archaeal sequence 2, reduced ~4% in length in bacterial sequences 3-5, or kept intact in the rest). The histogram shows the percentage of gaps in each column of the trimmed alignment.
Despite methodological problems and inconsistencies introduced by sequence analysis, the “canonical” bacterial rooting of the initial studies,81,82 which forced archaeal and eukaryal sequences to be sister groups to each other, was quickly endorsed by the microbiology community. 92 It has been accepted as fact despite cautionary alerts83,86,87 and substantial genomic evidence to the contrary (reviewed in Caetano-Anollés et al 93 ). While the canonical rooting of the Woesian 3-superkingdom ToL now populates numerous textbooks, a 2-superkingdom view of cellular diversification has been pronounced that trumps the canonical rooting by entailing an unlikely cellular fusion. 50 This view is now widely celebrated,90,92 despite of it stemming from unrooted phylogenies and being at odds with the history of structural domains and many other lines of evidence. In fact, the 2-superkingdom view has been also challenged on many grounds, from technical to biological,44,95 making the Woesian scenario for the global structure of diversified life far more likely.96,97
The use of the paralogous gene-indel rooting approach (and Weston rule) can be deceptive when genomic sampling is limited. Gene sets may provide discordant information because of homoplasy, including the effects of historical heterogeneities in gene makeup and global effects of horizontal gene transfer. These limitations can be mitigated by increasing the level of universality of phylogenetic statements, something that can be directly accomplished at the level of the character. The original promise of whole-genome biology 98 was to increase the levels of universality by providing comprehensive evolutionary information from entire repertoires of molecular traits (eg, genome, proteome, interactome). However, only few whole-genome evolutionary studies rendered rooted trees that would fulfill all tenets of evolutionary analysis. The iterative accumulation of homologies in paralogous protein-encoding genes was only recently stepped up orders of magnitude by focusing on entire families of genes at genome level. 99 In this study, single-nucleotide polymorphisms (SNPs) from whole-genome sequences of an obligate intracellular bacterial pathogen Coxiella burnetii were first used to create an unrooted phylogeny, which was rooted by identifying polymorphic duplicated regions and using them massively as paralogs for the application of Weston rule. This approach has not been applied yet to the rooting of the ToL. In contrast, the iterative accumulation of structural domains has been effectively used for more than a decade to root ToLs describing the evolution of proteomes (reviewed in Caetano-Anollés et al30,93). The rationale of the approach is straightforward and is grounded in simple cladistic principles. Domain structures spread by recruitment in evolution when genes duplicate and diversify, genomes rearrange, and genetic information is exchanged. A genomic census of the occurrence and abundance of structural domains in proteomes and their combination can therefore be used to build rooted trees describing the evolution of domains and proteomes.100-102 In these studies, the abundances of domains are encoded as Wagner ordered multistate characters (Figure 1), which are first used to build unrooted trees. These trees are then rooted most parsimoniously by polarizing character state changes with Lundberg and Weston rule. A similar approach that generates rooted phylogenies with the generality criterion uses a census of molecular functions defined by Gene Ontology (GO) definitions.103-105 The abundance of structural features of molecules has also been used to build rooted phylogenies, starting with an analysis of the structure of the large and small subunits of ribosomal RNA (rRNA) 106 but also focusing on highly informative and ancient smaller molecules such as tRNA, 107 5S rRNA, 108 and RNase P RNA. 109 In all of these cases, ToLs were consistently rooted paraphyletically in Archaea, suggesting this domain of life was the first diversified supergroup to appear in organismal evolution (reviewed in Caetano-Anollés et al 93 ).
Other recent attempts to root ToLs generated from whole-genome biology use rooting methods that are either technically flawed or invoke additional auxiliary assumptions that are difficult to justify. For example, Harish et al 13 used a census of structural domains (as in Caetano-Anollés and Caetano-Anollés 100 ) and custom asymmetric stepmatrices (seeFigure 1) that penalize gains over losses to generate trees rooted in Eukarya. As mentioned earlier, this approach violates the triangle inequality and is subject to numerous technical and conceptual problems.110,111 To make the problem of this approach explicit, Wheeler 15 uses the well-known NP-hard “traveling salesman problem” to illustrate how “non-metric distances can have unforeseen and sometimes bizarre effects.” He imagines a salesman that wishes to visit a collection of cities while minimizing travel time. The task is known to require considerable optimization effort. However, a decision to use non-metric distances makes a city have zero distance to all other cities, creating a “wormhole” in space-time that allows to reach all cities at zero cost. Such property can have dire consequences during tree searches for the recovery of a correct tree. Another ill-conceptualized approach is the use of pseudo-outgroups to root distance-based trees inferred by studying the frequency of l-mer sets of amino acids in proteins at the proteome level. 112 The ToL generated from compositional data was rooted in Eukarya by using randomized proteome sequences as outgroups. The assumption of randomness equating ancestrality is, however, unsupported or probably wrong, especially because protein sequence space and its mappings to structure are far from random. 113 More importantly, using these random pseudo-outgroups as taxa imply that a large fraction of modern proteins that had already evolved prior to the appearance of the last universal common ancestor of cellular life had to be random. The existence of a universal core of ancient protein domains with well-defined structures falsifies the auxiliary assumption. 114 Finally, the midpoint rooting approach was used to root network trees between Bacteria and Archaea built from gene families defined by reciprocal best BLAST hits. 115 In this study, the assumption of a molecular clock is complicated by the reticulations generated in the network analysis, which require complex optimization of path lengths in split networks.
Benefits and Emergent Properties of Phylogenomic Abundance
Cladistic methodologies that focus on genomic abundance, ie, the incidence of genomic features in a genome, benefit from the study of entire genomic repertoires and well-established methods of phylogenetic analysis. These genomic features must be evolutionarily conserved and may include paralogs of gene families, structural domains, domain combinations, and GO definitions of molecular functions. Genomic abundance makes explicit the iterative accumulation of homologies, which allows to fulfill the generality criterion and Weston rule. Recall that character abundance can be decomposed into separate instances of occurrence without impacting phylogenetic optimization, extending the breath of abundance to many kinds of data. Cladistic methodologies that focus on abundance are powerful. They take advantage of permanent advances in structural and functional genomics and better machine-learning and supervised computational methods. For example, the genomic census of structural domains or GO terms expands its breath with scientific exploration, increasing the explanatory power and universality of trees and networks. 103 Similarly, HMM libraries that are used to define taxa and characters (eg, structural domains) are permanently upgraded by the survey of atomic structures and appropriate experimental exploration of molecular functions. The timing is also perfect. Structural data are starting to accumulate exponentially as genome data did in the last decade enabling these explorations.
Phylogenomic analysis of abundance reveals emergent properties of evolutionary nesting and growth. In general, taxa with character state vectors showing overall low abundance levels populate the base of rooted ToLs, while taxa with larger abundance vectors appear later in evolution closer to the crown of the trees. We have shown that this general pattern does not result from a “small proteome attraction” artifact pushing low abundance to the base of rooted ToLs reconstructed from a census of protein domain structures in cellular organisms and viruses. 111 Instead, our study reveals that this pattern arises in evolution by retention of iterative homologs, which nest in the unfolding phylogeny and are used by Weston rule to root the trees. Note that during searches of tree space and prior to rooting, character change is optimized in the unrooted trees. This allows unrestricted gains and losses of domain occurrence or abundance throughout branches, 100 which are amenable to phylogenetic reconstruction. 116 Thus, character polarization plays no role in defining unrooted ToL topology, which by definition cannot be distorted by proteomic abundance levels (proteome size), ie, by a property of taxa and not individual characters changing in trees. 111
The direction of the hierarchical nesting patterns can be uncovered by studying alternative character polarization schemes with the Lundberg method. First, optimal unrooted ToLs are generated from the multistate ordered characters with Wagner optimization. A hypothetical ancestor is then attached most parsimoniously to the internode of the unrooted trees a posteriori with Lundberg optimization. The “standard” implementation of Lundberg, which complies with Weston rule of the generality criterion, sets all character states of the ancestor to unknown or “missing” (ancstate = ?) and proceeds to optimize attachment of the best ancestor to optimal trees. 47 Alternatively, arbitrarily defined ancestors can be optimally attached to the most parsimonious tree reconstructions. The resulting alternative Lundberg polarization schemes can be compared with the standard implementation to determine which rooting schemes are more parsimonious and less affected by homoplasy. Figure 6 shows a sampling of most parsimonious ToLs describing the evolution of cellular proteomes that were rooted with alternative Lundberg implementations. Trees were rooted with Lundberg using the standard (all-?) ancestor or each of 32 possible ancestors holding the same ancestral state for every character, ie, ancestors that assign state i to the entire character ensemble (eg, for an all-0 ancestor, i = 0 using command ancstate = 0). The lengths and ensemble retention indices (RI) for optimal trees were recorded. The length of a tree measures how parsimonious is the phylogenetic reconstruction. The RI tests both the fit of character data to a reconstructed tree and levels of homoplasy in the analysis. 117 An RI value of 1 implies perfect fit and absence of ad hoc assumptions of homoplasy. An RI value of 0 implies the tree fits data as poorly as possible and exhibits maximum instances of independent origin. The length and RI values of trees decreased monotonically when Lundberg ancestors with increasing values of i were used to root the trees. However, the standard, “all-0” and “all-1” ancestors produced the most parsimonious and best ToL reconstructions out of all possible Lundberg implementations. These trees were the shortest, had the highest RI values, and had identical topologies. They placed Archaea at their base. Note that the topological isomorphy and optimality of ToL reconstructions using the “standard” and “all-0” ancestor implementations of Lundberg for structural domains have been repeatedly observed in our laboratories for more than a decade. Maximum parsimony and the generality criterion of rooting consistently support the Archaea-first hypothesis. 93 Results have important implications for phylogenetic analysis of proteomes: (1) Proteome data significantly fit the model of ordered characters, accumulation, and retention of serial homologs and evolutionary growth in the nested lineages of the ToL; (2) optimal character polarization with standard and “all-0” ancestors and monotonic increases of tree suboptimality shows there is a tendency of growth of structural domains in proteomes, and not global tendencies of reductive evolution; and (3) this tendency of proteomic growth preserves the regular pattern of character state distribution in the ToLs that results from the nesting of serial homologies. Results also add to the long list of evidence in support of the evolutionary axiom of spatiotemporal continuity. For example, phylogenetic tracings of proteome size in ToLs derived from a universal biology of evolutionarily conserved protein folds and along historical multidimensional projections (see evolutionary principal coordinate [evoPCO] in the following) revealed a slowdown in innovation of the structural domain vocabulary. 111 It also revealed a hidden interplay between protein fold innovation and abundance. This interplay materialized in four regimes of allometric scaling reflected in a Heaps law of vocabulary growth. 111 These regimes explained increasing economies of scale in the evolutionary growth and accretion of kernel proteome repertoires, which resembled growth of human languages with limited vocabulary sizes, such as the Korean or Chinese languages (eg, Li et al 118 ). Results reconcile dynamic and static views of frequency distributions of protein domains that are consistent with the axiom of continuity that is cornerstone of evolutionary thinking and ToL reconstruction.

Rooting trees of life (ToLs) a posteriori with the Lundberg method. Unrooted ToLs were generated using ordered characters and Wagner optimization (see Figure 1C) from phylogenetic encodings of genomic counts of 1420-fold superfamilies of protein structural domains in 102 proteomes (dataset from Kim and Caetano-Anollés 114 ). Proteomes were from organisms with free-living lifestyles equally sampled from superkingdoms Archaea (red), Bacteria (blue), and Eukarya (green). Trees were rooted with Lundberg using the “standard” implementation (anscsate = ?) or with 1 of 32 possible ancestors holding the same ancestral state for every character. Character states describing genomic abundance levels were labeled in alphanumeric format from 0 to 9 and A to V. Trees lengths and ensemble retention indices (RI) were calculated for recovered trees. Lengths were described with a line graph with data points labeled with closed and open symbols if trees had topologies with paraphyletic or monophyletic basal superkingdoms, respectively. Colors described support for an Archaea-first or Eukarya-first evolutionary scenario of origin. Control experiments that generated rooted ToLs using ordered maximally connected characters and Fitch optimization still produced most parsimonious reconstructions rooted in Archaea when using standard and all-0 Lundberg ancestors. However, the monotonic decrease of lengths and RI values was not evident.
Parts and Wholes and the Ontology of Tree Building
Ontology embodies the naming and description of concepts and relationships that exist for an agent or community of agents interacting with their worlds. The agents are goal-oriented existing entities and abstractions grouping entities according to shared or distinct features. This definition is not distant from that of computer and information sciences, which consider ontologies as abstractions defining “representational primitives,” ie, the naming and definition of sets, properties, and relationships of entities. When entities are described within a framework of “systems theory,” 119 parts of systems (generally cohesive units, modules) and their interrelationships are named and their complexity defined both ontologically (pertaining to existence) and epistemologically (pertaining to knowledge). For example, machine-learning or supervised approaches can be used to classify structural domains or conserved sequence elements, which are part of the biological system’s whole, in this case the proteome of an organism. Similarly, gene ontologies in the GO database define a controlled vocabulary of gene or gene product attributes of molecular functions (mf), biological processes (bp), and cellular compartments (cc) that distill the molecular essence of life in an organism’s functionome. 120 These classifications describe systems with sets of parts that are finite. As long as the assumption that parts have been appropriately surveyed is appropriately justified, classifications of these kinds tend to attain the highest level of universality. Their evolutionary implications can be put to the test.
When reconstructing biological history, the overwhelming focus has been the use of biological systems (organisms) as taxa, dating back to the work of Haeckel. Generally, trees describing the evolution of parts have been generated to indirectly inform about the evolution of systems or to infer local statements of relationships of those parts (eg, trees of genes) (discussed in Caetano-Anollés et al 93 ). However, a phylogenetic data matrix can be transposed (by switching rows and columns) to generate trees of systems and parts from the same features of biological systems that are being studied. Figure 7 illustrates the transposition of phylogenetic matrices of occurrence or abundance of useful features, focusing on the study of molecular parts in molecules, protein domains in proteomes, and molecular functions in functionomes. Remarkably, trees of parts have the potential to better explain evolutionary relationships of systems for several reasons. First, parts allow exploration of those that are shared or are unique to systems, helping find relationships that exist between them, and in doing so, explaining (with minimum bias) the systems per se. For example, viruses and cellular organisms share and harbor unique structural domains, the history of which can describe the history of the viral and cellular systems, independent of how they are defined or considered (eg, nonliving or living entities akin to cellular organisms 121 ). Similarly, the concatenated alignment example of Figure 5 shows how the standard sequence analysis of systems (organisms) forces the trimming of phylogenetically informative features, including anticodon-binding and tRNA-binding domains or structurally important segments of domains, some holding deep organismal history. Trees of sequence parts can be used to generate history of systems that respects the historical heterogeneity of sequence makeup which is trimmed in concatenated sequence alignments (unpublished data). Second, trees of parts can help circumscribe biological systems by testing the strength of homology statements imparted by system-describing characters (eg, Kim and Caetano-Anollés 114 ). Finally, and more importantly, the evolutionary study of parts diminishes the serious problem of violation of character independence that challenges phylogenetic reconstruction 93 and at the same time tests Kluge auxiliary principle. The assumption of independence of systems used as characters to build trees of parts can be better justified than the independence of parts used as characters to build trees of systems, as parts are by definition (and ontologically) interacting components of systems that are not independent from each other. While systems can depend on other systems, their interactions are of higher level and are above the definition of the character-taxa set being studied.

Thanks to Weston generality criterion and as showcased in Figure 3, a rooted tree describing the evolution of characters informs construction of a rooted tree describing evolution of taxa, whether taxa represent parts (or systems) and characters represent systems (or parts), respectively. Thus, the interplay of trees derived from any matrix describing systems and parts makes explicit Weston rule and the task of rooting phylogenies. Figure 8 shows examples of the tracing of character state change onto the branches of a universal ToL that describes the evolution of cellular and viral proteomes. In the presence of significant vertical phylogenetic signal, domains that appear early in the protein world (with relative ages approaching 0) have higher chances of spreading widely through the lineages of the ToL while those that appear late are usually confined to smaller sets of organismal taxa that are increasingly restricted to specific locations in the tree closer to the crown. This pattern is plainly evident in a phylogenetic data matrix and the trees of parts and wholes that are reconstructed from it. A character tracing exercise of changes in domain abundance along branches of a ToL describing evolution of proteomes shows indeed how the oldest domains contribute to establishing basal and widely spread phylogenetic relationships that comply with Weston rule of iterative accumulation of homologies (abundance) in the nested lineages of reconstructed trees.

Unfolding Weston rule by building trees of parts and wholes from abundance of structural domains in proteomes. A phylogenetic data matrix with columns describing 442 structural domains (parts) defined at fold superfamily level and shared by all supergroups (viruses and Archaea, Bacteria, and Eukarya) and rows describing 5080 proteomes (wholes) was used to build a tree of domains, which was used to order domains in the matrix according to evolutionary age (derived directly from the highly unbalanced tree). The data matrix in heat map format (red-blue describing domain presence-absence) and the tree of domains are shown one above the other in the left. A plot of the spread of domains in proteomes (f) plotted against domain age (nd, described in text) was overlaid over the heat map. Widely distributed domains shared by cells and viruses are indexed with SCOP ccs. The yellow smoothed curve describes the relationship as determined by the LOWESS method (100 iterations, q = 0.07). A ToL reconstructed from genomic abundances of domains in a proteome subset (368 genomes) is shown in the right with character state reconstructions for the three oldest and most widely distributed domains (c.37.1, e.8.1, and c.55.3) traced along the branches of the tree of proteomes. Arrowheads indicate the most basal character state change contributing to Weston rule. Note how the oldest domains are the most widely distributed in proteomes and how their abundances change along the most basal branches of the ToL. The sparse distribution of domains in viruses is also noteworthy, with exceptions in large dsDNA viruses. ToL rooting is, however, enabled by the nesting of character state change in the tree that stems from decreasing pattern of domain distribution in proteomes with evolutionary age (see the f vs nd plot). Rooting unfolds despite the very sparse viral distributions.
Building Organismal History From Trees of Molecular Parts
Trees of domains, such as the one described in Figure 8, have been reconstructed from phylogenomic data summarizing ~11 million proteins of 5080 proteomes and holding significant phylogenetic signal. 121 Their branchings are well supported, especially at their base. Their comb-like shape suggests recurrent episodes of punctuation and gradual appearance of structural domains in protein evolution. The highly unbalanced topologies of the trees enable the calculation of a relative age or node distance (nd) for each and every taxon, ie, each and every structural domain examined. This is accomplished by simply counting the number of nodes that are present in a path from the node that roots the tree to each taxon leaf and dividing that number by the maximum number of nodes in the paths. The gradual evolutionary recurrence manifests in molecular clock-like behavior, a linear correlation between nd and geological time calibrated by the use of fossil, biomarker, and other evidence. 122 The recurrence has been explained with global dynamic models describing the evolution of domains, fitted with phylogenomic data. 123 The models suggest that proteins explore the space of possible molecular structures through coarse-grained discoveries that undergo fine-grained elaboration. These repeating (self-similar) patterns of molecular diversification are typical of expanding symmetry in the fractal behavior of multi-layered systems.
Given a historical account of evolution of parts, the repertoire of parts defines a repertoire of ages of those parts that can be mined to generate historical accounts of systems. For example, Nasir and Caetano-Anollés 121 recently developed a metric multidimensional scaling approach to study the evolution of the proteomes of cellular organisms and viruses. This evoPCO analysis method (1) combines the power of cladistic and phenetic approaches, (2) extends the ability of multivariate statistical analyses to summarize high dimensional data (eg, for microbial ecology 124 ) to problems of deep evolution, and (3) helps minimize violation of phylogenetic character independence. Its application to proteome evolution takes advantage of the fact that proteomes are made up of structural domain parts, each of which has its own age of origination and its own nd directly derived from a tree of domains. As our modeling exercise has shown that domain structure (suitably defined) arises through course-grain explorations, the use of domain occurrence rather than abundance appropriately surveys the parts that will be contributing ages to individual ages of the cellular or viral proteome systems. Operationally, a matrix of domain abundances gij, describing i domains and j proteomes, is simultaneously transformed into a domain occurrence matrix (similar to that of Figure 8) and a domain age matrix by multiplying the occurrence of each domain by the reverse of its corresponding age

where ndi represents the age of domain i and Iverson brackets transform abundances into occurrences. The reverse age transformation ensures both that the oldest domains (of age nd = 0) contribute appropriate age to the multidimensional temporal space and that domain absences (gij = 0) do not, as absences are domains that have not yet materialized in evolution. The method also assumes that the age of a domain is the age of the first evolutionary appearance of that domain. Euclidean distances reflecting pairwise age dissimilarities between proteomes were used to directly calculate principal coordinates describing maximum variability in temporal data. Figure 9 shows phylogenetic dissimilarities displayed in a low-dimensional (three-dimensional [3D]) temporal space describing how domains (defined at fold superfamily level) contribute to proteome history. 121 The three most significant loadings of the evoPCO method, which account for 85% total variability, reveal four clearly separate temporal clouds of proteomes, each representing a supergroup, Archaea, Bacteria, Eukarya, and viruses. A reconstructed proteome of the last universal common ancestor of cells 114 served as time reference in the multidimensional temporal space, showing that viruses originated prior to cellular superkingdoms and that the rise of Archaea preceded that of Bacteria and Eukarya. It also showed the early evolution of RNA-segmented viruses (Lassa virus) and the late appearance of giant viruses (Mimiviruses, Megaviruses, and Pandoraviruses). Thus, the evoPCO projection of a multidimensional space onto a 3D temporal space enables a unique and powerful visualization of deep evolutionary relationships. The approach is general and can be applied to any kind of phylogenetic character and any kind of taxa, as long as characters are quantifiable parts describing taxa.

The origin and evolution of proteomes inferred from multidimensional scaling analysis of domain age (from Nasir and Caetano-Anollés 121 ). (A) Timeline describing the evolution of structural domains defined at SCOP family level responsible for the modern protein world. The timeline was derived directly from a tree of domains. Ages (nd) are given in geological time measured in billions of years (Gy) according to a molecular clock of folds, with time flowing from left to right. The three evolutionary epochs of the protein world, “architectural diversification” (Epoch 1), “superkingdom specification” (Epoch 2), and “organismal diversification” (Epoch 3; see definition in Caetano-Anollés et al 89 ) are indicated with different color shades. Some fundamental structural and functional discoveries are identified with dots along the timeline. B. An evoPCO analysis plot portrays in its first three axes the evolutionary distances between cellular and viral proteomes. The percentage of variability explained by each loading is given in parentheses on each axis. The reconstructed proteome of the last common ancestor of modern cells 114 was added as a reference to infer the direction of evolutionary change.
Effects of Organismal Lifestyles, Problematic Taxa and Character Ontology in Tree Reconstruction
The definition of taxa is particularly problematic for ToL reconstructions. The concept of the “holobiont,” the collective of a host and its symbionts,75,76 has blurred the definition of an organism. 125 Its ubiquity has challenged the “biological species” concept of unit of biological organization 126 while accommodating “multilevel selection theory.” 127 Holobionts are ill-defined collectives that are highly dynamic. Examples include humans and their microbiomes or corals and their associated zooxanthellae, endolithic algae and bacteria. Their “hologenomes” encode molecular repertoires that are themselves ill-defined and highly dynamic. This complicates the definition of taxa and genomic repertoires used for their evolutionary analysis. 120 Holobionts and hologenomes prompt (1) revisiting the idea that selection operates at multiple levels in the nested hierarchy of life, (2) examining what is “organism” and what is “environment” when defining systems, and (3) re-defining cohesiveness as argument for definition of systems and taxa.
Besides symbiosis, obligate parasitism also poses problems, especially when the genomic makeup of parasites is highly reduced.128,129 With exceptions, 130 many organisms that engage in obligate associations harbor minimalistic genomes. For example, Cand. Tremblaya princeps is not an independent organism, rather an organismal consortium. 131 With a genome that encodes only 55 universal domain superfamilies, it relies on its host (Planococcus citri) and its endosymbiont (Cand. Moranella endobia) to synthesize essential metabolites. 132 Similarly, Cand. Nasuia deltocephalinicola is an obligate endosymbiont of leafhoppers. It harbors the smallest known bacterial genome, 133 encoding only 53 universal domain superfamilies. These hologenomes with extreme proteomic outliers arise from relatively modern genomic losses, exchanges and recruitments likely resulting from complex trade-off relationships that complicate the dissection of their evolutionary origin. They represent “problematic” taxa that should be excluded from phylogenetic analysis to avoid biased tree reconstructions. 111 For example, when building ToLs from structural phylogenomic data, the exclusion of organisms that do not engage in free-living relationships avoids many pitfalls. 134 Their inclusion, however, can be justified if the obligate parasitic or symbiotic mode is a hallmark of entire groups of taxa that are being sampled. Such is the case of viruses, which, as a group, harbor life cycles with strict dependence of the host.111,121 In all cases, taxa should be sampled randomly, equally, and densely from each major group of organisms being analyzed for reliable tree reconstruction.135,136
The definition of taxa in a ToL is even more problematic when features of individual molecular parts of organisms (eg, 16S rRNA or protein-encoding genes) are used as characters to study their evolution. For example, when a ToL is reconstructed from sequence alignments of genes, the sequences are considered to represent organismal taxa and the ToL is said to describe the evolution of life. However, the “sequence-equals-organism” assumption is a “leap of faith,” since characters describe properties of molecular sequences not organisms. In the case of Figure 5, the “advanced” model of sequence evolution (PROTGAMMALG) used by Spang et al 90 to build a ToL from a concatenated sequence alignment of universal genes simply describes the process of replacement of amino acids in the sequence sites of a handful of sampled proteins. It does not describe processes of (1) diversification of molecular structure, which likely involves a multiplicity of structural domain recruitments (Figure 5B), (2) evolutionary constraint of structural domains induced by their functional sites, 137 and (3) selection acting at multiple levels of the nested hierarchy of life responsible for organismal diversification that the “sequence-equals-organism” paradigm implies. Furthermore, while all proteins in the alignment of Spang et al 90 are considered universal, (1) not all of their structural domain components are shared by all sampled organisms, (2) not all universal domains hold the same lower level structural motifs in the molecules sampled, and (3) not all aligned sequence site contribute equally to that universality.91,95 This questions the “sequence-equals-organism” ad hoc of universality that interprets characters and taxa.
Hennig 2 definition of taxa was originally associated with the “semaphoront” concept, the concept of being a “character bearer.” A semaphoront is an organism at a point of time in its development and evolution harboring a complete set of characters describing it, ie, its “holomorph.” The concept is relevant for definition of taxa, as semaphoronts are dynamic entities composed of holomorphs that are themselves dynamic. The ontological complexity of the semaphoront-holomorph relationship can be dissected with computer-aided ontologies for semantic organization, but the challenge of doing so is significant for both phenomics and evolutionary biology.138,139 This brings us back to the problem of parts and wholes of systems.
Conclusions
There are multiple explanations for our inability to provide a clear picture of organismal diversification (besides apriorism and ad hocness), including biases introduced when rooting trees with outgroups, insistence of using organismal systems as taxa, disregard for violations of character independence, and inability to recognize fractality (similar patterns at different scales) and multi-level selection when defining units of evolution in biology. One solution to the problem is the use of multidimensional scaling methods to study the evolution of biological systems through the age of their component parts, with ages being directly drawn by building rooted trees of parts. The use of the generality criterion to root and visualize the progression of innovation of biological parts takes full advantage of what we have learned in more than half a century of retrodiction research. There should be no assertions or assumptions about biological processes in a tree searching algorithm, nor should these assertions or assumptions be used to root the reconstructed trees. Processes should be inferred following an analysis of data, not built into the analysis. For example, application of the generality criterion confirms the belief that structural domains and molecular functions that are ancient are both abundant and widely distributed in nature. Similarly, the generality criterion confirms the idea that molecules and molecular parts with constrained numbers of conformations are older, lending support to the intuition that molecular conformations must be evolutionarily optimized to exist long enough to hold functions.
Farris 140 recognition that character polarity is unimportant prior to phylogenetic reconstruction with the Wagner algorithm is also fundamental and helps disentangle the tree optimization problem from the tree rooting problem. An optimal unrooted tree can be selected from the set of all possible trees by tracing character state change along its branches, fulfilling an optimality criterion for evolutionary change, and minimizing homoplasy. Shared and derived character states (synapomorphies) will group taxa in the unrooted trees in the absence of character polarization and knowledge of which is the plesiomorphic (ancestral) or the apomorphic (derived) character state. Once the tree is rooted, shared and derived states are clearly identified and each homology statement can then be put to the test. Farris initial recognition of building unrooted trees a priori implied that parsimony (or other optimality criteria) was the central epistemological criterion for phylogeny reconstruction, especially because an unrooted tree has many possible roots (equal to the number of branches it holds) that are all equally parsimonious. However, while the length of the most parsimonious trees is unaffected by the position of the root, rooting impacts the homology statements of the unrooted trees.42,140 This fact is made evident by the Lundberg optimization method of attaching most parsimoniously outgroup nodes that are compliant with Weston rule. Thus, “The length of a tree is unaffected by the position of the root but is certainly not unaffected by the inclusion of a root,” 141 and consequently, parsimony plays an equally important epistemological role in the rooting of trees. We note, however, that both tree reconstruction and rooting benefit from the ontology of reciprocal illumination, showing that ontological and epistemological frameworks are tightly integrated.
We end by noting that evolutionary conservation implies an ability to differentiate transformational homologs, the ancestral from the derived state. As these states are causally related, the plesiomorphic state establishes a statement of origin in the phylogeny, which tests the initial homology statement of phylogenetic memory. If change between transformational homologs is too fast or too slow along phylogenetic branches, reliable plylogenetic signal will be difficult to extract from the biological features that are being studied. This decreases confidence in the phylogeny, including its associated rooting statement, weakening the phylogenetic test. For example, a focus on molecular sequences imposes unsurmountable burdens to finding reliable roots, even when sequence sites are selected that are highly conserved. This stems mostly from meager understanding of the sequence-to-structure mapping that is responsible for the function and stability of molecules. Thus, evolutionary conservation is a fundamental aspect of the retrodiction equation but its ultimate causes remain mysterious. When transformational homologs are many, such as static ordered characters describing abundance in biology, the multi-causal relationship between states helps both the tree optimization and the tree rooting problems. This is particularly so when the iterative process of evolutionary accumulation is slow, such as is the case of structural domain evolution. 123 In these cases, the biological accretion process acts as a “digital buffer” capable of providing power amplification to the phylogenetic signal. Thus, a focus on parts and their abundance in biological systems may provide new avenues of evolutionary exploration.
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Computational biology is supported by grants and computer allocations from the National Science Foundation (OISE-1132791), United States Department of Agriculture (ILLU-802-909 and ILLU-483-625) and a NCSA Blue Waters to GCA, the Higher Education Commission Start-up Research Grant Program (Project No. 21-519/SRGP/R&D/HEC/2014) to AN, and the Collaborative Genome Program (20140428) funded by the Ministry of Oceans and Fisheries, Korea to KMK. DCA is recipient of NSF postdoctoral fellowship award 1523549.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
All authors contributed to the planning, conception and interpretation of the work and materials presented in this review. The lead author wrote the manuscript with the help of all co-authors.