
Abstract
β-lactamases, the enzymes responsible for resistance to β-lactam antibiotics, are widespread among prokaryotic genera. However, current β-lactamase classification schemes do not represent their present diversity. Here, we propose a workflow to identify and classify β-lactamases. Initially, a set of curated sequences was used as a model for the construction of profiles Hidden Markov Models (HMM), specific for each β-lactamase class. An extensive, nonredundant set of β-lactamase sequences was constructed from 7 different resistance proteins databases to test the methodology. The profiles HMM were improved for their specificity and sensitivity and then applied to fully assembled genomes. Five hierarchical classification levels are described, and a new class of β-lactamases with fused domains is proposed. Our profiles HMM provide a better annotation of β-lactamases, with classes and subclasses defined by objective criteria such as sequence similarity. This classification offers a solid base to the elaboration of studies on the diversity, dispersion, prevalence, and evolution of the different classes and subclasses of this critical enzymatic activity.
Introduction
The increasing amounts of genomic data produced by next-generation sequencing technologies made β-lactamases (BLs), the enzymes responsible for the irreversible inactivation of β-lactam antibiotics, one of the most numerous families of proteins studied to date. 1 First described by Abraham and Chain, 2 BLs can be found in pathogenic or commensal bacteria, isolated from humans or varied environments. 3 Due to its great medical importance and clinical impact, several groups directed efforts in the development of a proper BL classification scheme, usually based on functional or structural criteria.4–6
Function-based classification is achieved using experimental data to link the enzyme to its clinical role.6,7 The determination of these parameters for a large number of BLs, however, may be relatively costly and time-consuming. Also, they do not generate sequence data, essential for studies involving molecular evolution. 8
Currently, the most widely used classification scheme for BLs is the Ambler structural classification, which is based on sequence similarity, and separates BLs into 4 classes: the classes A, C, and D of serine-β-lactamases (SBLs) and the class B of metallo-β-lactamases (MBLs).4,9,10 Class B is further divided into subclasses B1, B2, and B3, using sequence conservation data. 11
Despite SBLs and MBLs are able to break amide and ester bonds (EC 3.5.2.6), they belong to 2 distinct protein superfamilies that do not share a common ancestor. 12 Considering SBL’s tertiary structures, they are similar enough among themselves to be considered homologous, 5 whereas the differences between their primary structures and catalytic mechanisms justify their division into classes A, C, and D. 13
Experimental data indicate that the 3 subclasses of MBL should not be treated as equally separated groups. Subclasses B1 and B2 have detectable sequence similarity between them but not with B3, 14 and structural evidence strongly suggest different Most Recent Common Ancestor between the group formed by subclasses B1 and B2 and the subclass B3. 15
Based on this structural information, a reorganization of BL at 4 hierarchical levels was proposed by Hall and Barlow. 5 In this scheme, the former subclasses B1 and B2 were merged and renamed as class MB, whereas subclass B3 was renamed as class ME. Thus, the 5 BL classes (third classification level) are SA, SC, SD, MB, and ME, and subclasses MB1 and MB2 represent the fourth and last hierarchical level. 5
Moreover, recent studies employing phylogeny and amino acid-based sequence similarity networks showed that classes SA and SD could be further divided into 2 different groups, suggesting new BL subclasses.1,16 Indeed, the current molecular classification of BLs does not represent its actual sequence diversity, and there is yet no precise definition of various classes and subclasses.1,5,16
Profiles Hidden Markov Models (HMM) and sequence clustering using similarity underlie the workflow proposed here. It is based on a previous hierarchical scheme which reflects the distinct evolutionary origins of SBLs and MBLs. 5 Application of this scheme to 2774 assembled bacterial genomes provided improvements in BL annotation and in the knowledge about BLs distribution among bacteria phyla, confirming previous studies suggesting new BL subclasses.1,16 Finally, our results propose the existence of a new BL class with fused domains and extended action spectrum.
Methodology
Data collection and preparation
On March 22, 2016, an online search for BL structures using the EC number (3.5.2.6) in the National Center for Biotechnology Information (NCBI) Structure Database 17 returned 516 entries. Records with the word “mutant” in the description were excluded. The PDB (Protein Data Bank) IDs were retrieved and used for downloading PDB files and their corresponding FASTA files from RCSB PDB. 18 The data were filtered using the following criteria: duplicate atoms positions were removed, the resolution of the structure should be less than 3 Å, and only monomers or the chain A from homomultimers were used.
A Non-Redundant Beta-Lactamase Dataset (NRBLD) was constructed with protein sequences retrieved from 7 different antibiotic resistance databases. Four databases are specific for BLs: (1) Comprehensive Beta-lactamase Molecular Annotation Resource (CBMAR, downloaded on August 2015), 19 (2) The Institute Pasteur Database (downloaded on October 2015), 20 (3) DLact Antimicrobial Resistance Gene Database (downloaded on October 2015), 21 and (4) Lactamase Engineering Database (LacED, downloaded on August 2015). 22 The Metallo-Beta-Lactamase Engineering Database (MBLED, release 1.0) is the only specific for MBLs. 23 The remaining 2 databases, Comprehensive Antibiotic Resistance Database v1.0.0 (CARD) 24 and Resfams v1.2, 3 have protein sequences related to antibiotic resistance in general. To recover only BL sequences, we search for those with the term “bla” and “beta” in the headers. Identical sequences have been removed using CD-HIT 25 at a 100% identity threshold. The following methodology steps are summarized in Figure 1.

Main conceptual steps of the workflow.
Clustering curated BL structures and sequences
The clustering assays were made using BL structures and their corresponding amino acid sequences from PDB, in an attempt to reproduce the BL classes and subclasses proposed by previous works. The MaxCluster program 26 was used to cluster the structures based on root mean-squared deviation and hierarchical clustering, applying 3 tests: single, average, and maximum linkage. The BLASTClust v2.2.26 program 27 was used for the hierarchical clustering of the amino acid sequences, which performs a single linkage type clustering based on pairwise matches found by BLAST. Different threshold values of “BLAST score density” (BLAST score divided by the alignment length) and “minimum length coverage” were tested. An E value of 1E−05 was used.
Building a profile HMM for each BL class
The profiles HMM were constructed using the clusters of sequences corresponding to the 5 BL classes. 5 Because these sequences came from proteins with a resolved structure, they were considered reliable to be used to construct the profiles. Sequences from each cluster were saved in separate multi-FASTA files. Identical sequences were removed using CD-HIT 25 at a 100% identity threshold. Sequences in each file were aligned using MUSCLE. 28 Profiles HMM were built from each alignment using the program hmmbuild from the HMMER package v3.1b2. 29
Calibration and validation of the profiles HMM
The profiles HMM generated were used against the protein sequences at NRBLD and superfamilies 3.40.710.10 (DD-peptidase/BL like) and 3.60.15.10 (Metallo-BL like) from Protein Structure Classification Database (CATH) (downloaded on March 17, 2016). 30 Hmmsearch from the HMMER package v3.1b2 with an E value of 1E−05 was employed. 29
The Class profile Specificity index (CpSp) evaluates whether each profile identifies a unique group of sequences. CpSp was calculated by dividing the number of NRBLD sequences identified exclusively with a given profile (Ne) by the total number of NRBLD sequences recovered by it (T), including intersections with others profiles results [CpSp = (Ne/T)*100]. Profiles with CpSp below 100% were calibrated. Sequences from other classes that should not be identified were used as “negative training sequences,” following the HMM-ModE protocol. 31 After this, the hmmsearch Gathering Threshold (GA) parameter was used, substituting the E value.
A total of 851 amino acid sequences of DD-peptidase/BL-like superfamily downloaded from CATH 30 were used to construct an unrooted Maximum Likelihood phylogenetic tree in MEGA-CC v7.0.18, 32 using the Jones-Taylor-Thornton model, partial deletion for gaps/missing data treatment (95% site coverage cutoff) and 500 bootstrap replicates. Using in-house Perl scripts, the BL sequences were manually labeled with their respective class, and the clade node of each BL class was identified. The sequences in these clades were allocated in corresponding multi-FASTA files.
Graphics were created with all the sequences retrieved by each profile against the DD-peptidase/BL superfamily with their respective HMM bit score. These results were used to calibrate the profiles. A new hmmsearch parameter of HMM bit score threshold was established to separate true BL from other sequences in the superfamily. The Function Specificity (FSp) index verifies whether all sequences retrieved by a profile have the BL activity already described. For this, the number of superfamily sequences identified by a profile that has BL activity (Nbl) was divided by the total number of superfamily sequences identified by the profile (T) [FSp = (Nbl/T)*100].
A total of 144 sequences are attributed to the Gene Ontology (GO) molecular function “BL activity” (GO:0008800) in the SwissProt database (downloaded on March 2, 2016). 33 The validation index Function Sensitivity (FSe) evaluates the ability of all profiles together to identify the sequences annotated with the BL function in SwissProt. To calculate this index, the number of identified SwissProt sequences attributed to the BL GO term (Ngo) was divided by 144 [FSe = (Ngo/144)*100].
Validation of thresholds used to form BL subclasses
The results obtained with the curated PDB data set were used as a basis to group the NRBLD sequences into subclasses. The BLs from PDB were added to NRBLD, and the profiles HMM were used against them.
In order to reproduce BL subclasses, different thresholds of “minimum length coverage” were tested to cluster the sequences in each class, together with the “BLAST score density” thresholds previously established using the PDB sequences. The sequences in each cluster were annotated using the BLASTP 34 best hit (v2.2.28) against the nonredundant NCBI protein database. 17
The size of specific domains of BLs was used to stipulate a minimum size necessary for the enzyme to be functional. 35 The Pfam 36 models most common in MBL (PF00753), class SA (PF13354), and class SC (PF00144) have a length of 197, 202, and 330 residues, respectively. A total of 214 residues domain have been attributed to class SD enzymes. 37
The clusters formed were categorized as follows: (1) clusters corresponding to those obtained in the hierarchical classification of functionally characterized BLs (PDB sequences) and (2) clusters containing sequences that do not fit into the previous established similarity and coverage thresholds.
Comparison of the improved profiles HMM with Pfam profiles and BL motifs
To test the efficiency of our profiles HMM in identifying and classifying BL sequences, these were compared with profiles available in the Pfam database 36 and with motifs specific to BL classes from different sources.
The profiles HMM from Pfam 36 were used to search for BL sequences from PDB, and the CpSp indexes were calculated accordingly. Seven Pfam profiles (PF00144.22, PF13354.4, PF00753.25, PF12706.5, PF13483.4, PF14597.4, and PF16661.3) were downloaded on December 2016.
Motifs specific for BL classes obtained from the literature were evaluated for their ability to distinguish between classes and subclasses. An in-house Perl script was developed to identify the motifs in the BL sequences from PDB.
Identification and classification of BLs in fully assembled genomes
The workflow proposed here was applied to identify and classify BL sequences present in 2774 bacterial strains with fully assembled genomes that were deposited in NCBI. 17 A genome was defined as the complete set of chromosome and plasmids of each strain. The protein sequences present in each genome were downloaded (June 2016). Using the profiles HMM, the BL classes were formed, which were then separated into their respective subclasses applying the BLASTClust program 27 with the “BLAST score density” and “minimum length coverage” thresholds set in the previous step. The taxonomic information of each sequence was determined using the Genome Online Database (GOLD) 38 and in-house Perl scripts.
The scripts, profiles HMM, and instructions required to apply the workflow presented here, in addition to the data used for the searches, are available at https://github.com/melisesilveira/betaLactamase-classification.git.
Results
Clustering curated BLs structures and sequences
In total, 509 PDB structures and their respective sequences were used in the clustering and in the construction of profiles HMM. Of these, 208 were monomers and 301 were homomultimeric proteins.
Clustering of the curated set of structures using single linkage was consistent with the BL classification as proposed by Hall and Barlow, producing the same 5 classes (Figure 2). 5 Average and maximum linkage resulted in 7 and 18 clusters, respectively, and were not further used.
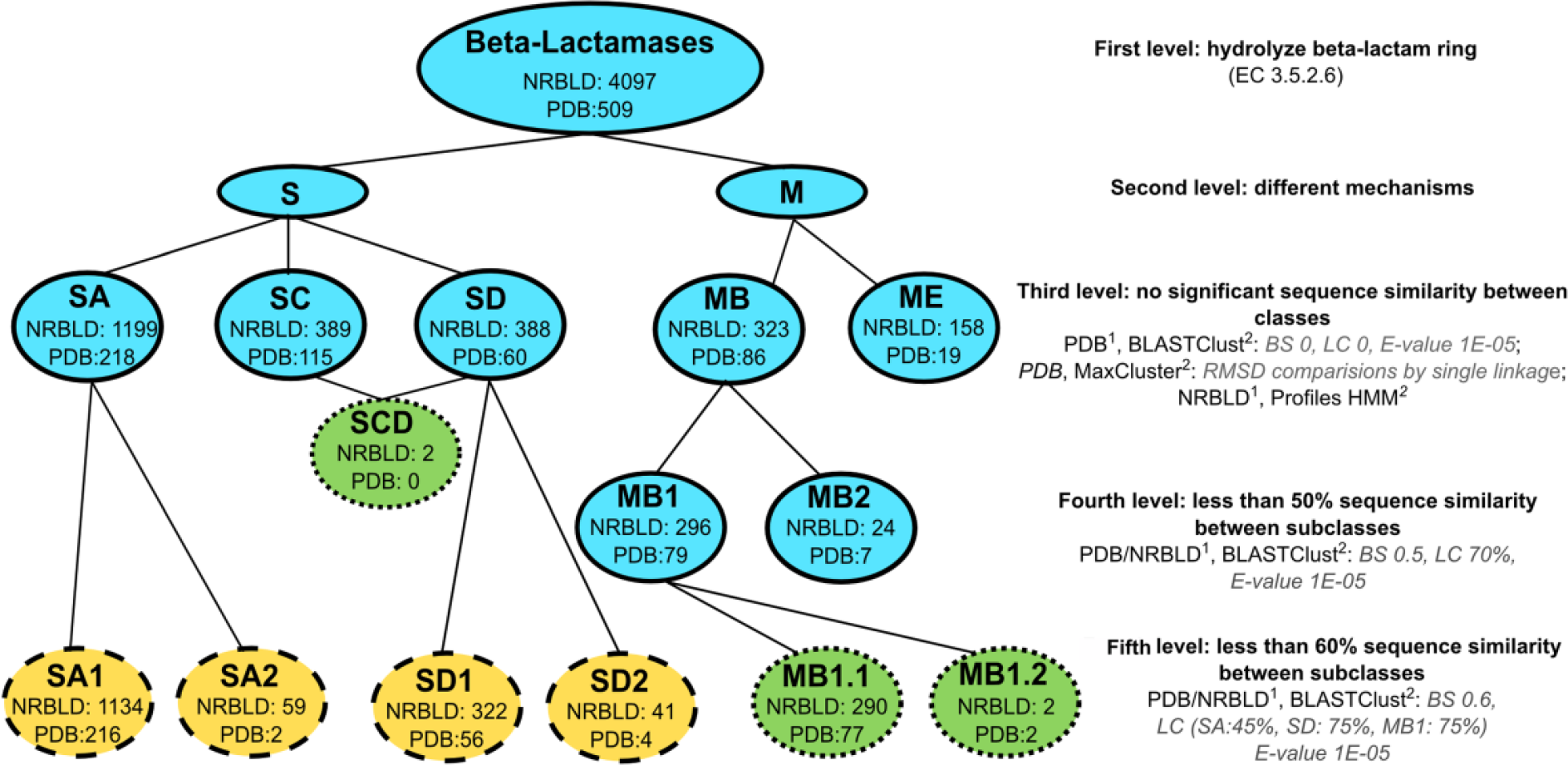
Hierarchical classification of β-lactamases.
The PDB sequences clustering applying “BLAST score density” thresholds of 0 and 0.5 were also consistent with the Hall and Barlow’s classification scheme, 5 producing the 5 BL classes and 2 MB subclasses, respectively. Using a “BLAST score density” threshold equal to 0.6, the subclasses proposed in previous works for classes SA and SD were reproduced,1,16 as well as the division of MB1 into 2 groups (Figure 2). The length coverage did not influence these results, and therefore a “minimum length coverage” of zero was chosen.
Separate clusters, one composed by 2 BL TEM-1 fused to a maltose-binding protein and another with 5 Escherichia coli penicillin-binding protein 5 (PBP5), were created in all clustering assays and were excluded because they are not true BLs, being wrongly associated with EC 3.5.2.6 in PDB. All sequence and structural clustering results are presented in Tables S1 to S3.
Building, calibration, and validation of the profiles HMM
Profiles HMM were built for each of the 5 clusters created using curated PDB sequences. These profiles were calibrated and validated using 3 sets of sequences: (1) NRBLD, our nonredundant protein set containing 4097 sequences; (2) the DD-peptidase/BL- and MBL-like superfamilies, with 851 and 399 protein sequences, respectively; and (3) SwissProt/UniProt 33 database, with 550 552 sequences. The profiles for the SBL and MBL classes were analyzed separately as these 2 groups belong to different CATH superfamilies. The data and profiles HMM are available at https://github.com/melisesilveira/betaLactamase-classification.git.
The profiles for classes SA, SC, and SD were searched against NRBLD recovering 1292, 1121, and 396 sequences, respectively. The profile of the class SA was 100% specific for the class (CpSp), whereas the profiles of the classes SC and SD were 99% specific. Two sequences were identified by both profiles, BL LRA-13 (ACH58991.1) and an enzyme annotated as “class C BL” of Janthinobacterium sp. (WP_008451281.1). Both have domains of SC and SD classes, which led us to propose a new class, SCD, composed of BLs with fused domains (Figure 2).
The profiles for MBL classes recovered 527 (MB, CpSp = 84%) and 612 (ME, CpSp = 86%) NRBLD protein sequences, 84 of which were identified by both profiles. Therefore, the profiles were optimized, and after the calibration they recovered 323 (MB) and 159 (ME) sequences, without intersections, reaching 100% CpSp.
To compare the calibrated and noncalibrated MBL profiles, they were tested against the 598 MBL protein sequences from the MBLED database. 23 Initial profiles identified 440 (MB, 85% CpSp) and 222 (ME, 70% CpSp) sequences, which represents 99.8% of the database. However, calibrated profiles identified 424 (MB, 100% CpSp) and 164 (ME, 100% CpSp) sequences, representing 98.3% of the database. The remaining 1.7% was composed of protein fragments ranging from 75 to 131 amino acids, meaning that the calibrated profiles were able to identify all the complete BL sequences.
The phylogenetic relationships of the SBL superfamily proteins showed a few sequences with no BL activity inserted into BL clades (Figure S1). The penicillin-binding proteins A (PBP-A) are in the inner branches of class SA and have a structure very similar to the BL PER-1 (subclass SA2) but do not have BL activity. 39 The regulatory proteins BlaR1 (and its cognate MecR1) are in inner branches of the class SD. Their extracellular domains are phosphorylated by β-lactams and, consequently, these proteins regulate resistance to these antibiotics in Staphylococcus aureus. 40
Initially, the profiles HMM for SBLs identified these sequences and others outside the BLs clades (all non-BLs). However, based on their HMM bit score values, a total of 75 non-BL sequences could be excluded (Figure 3). The separation of true SD2 BLs from BlaR1 proteins in this step was not possible because the HMM bit score of the single structure available of this subclass present in the CATH database 30 (OXA-1) was very close to some BlaR1 proteins (Figure 3). Additional tests have shown that when other variants of this subclass are included, their scores are smaller than those of some BlaR1 sequences. This can be explained by the structural homology of the extracellular sensor domain of BlaR1 to BLs from subclass SD2. 41

Plot of the 851 DD-peptidase/β-lactamase–like superfamily (CATH 3.40.710.10) sequences.
The enzymes ClbP and Pab87 are associated with the BL EC number in the PDB and share a significant similarity between their active sites with the BLs from class SC. 42 However, they can be separated from the class SC BLs by both phylogeny (Figure S1) and HMM bit score (Figure 3). Sequences in each BL clade and their respective HMM bit score are shown in Table S4.
HMM bit score thresholds of 120, 430, and 120 were defined for classes SA, SC, and SD, respectively (Figure 3). After the utilization of these bit scores, the FSp was equal to 100%, 100%, and 81%, for classes SA, SC, and SD, respectively. These thresholds retrieved 1199, 389, and 388 sequences from NRBD, respectively.
After the calibrations, all BL profiles together retrieved 132 proteins from SwissProt 33 : 82, 10, 24, 15, and 1 sequences with the profiles for classes SA, SC, SD, MB, and ME, respectively. In all, 125 out of the 144 sequences annotated with the BL GO term in this database were identified by the profiles (87% FSe). The remaining 19 sequences not identified as BLs presented one of the following annotations: BL fragments, BL-like protein, DacA carboxypeptidase, hydroxyacylglutathione hydrolases, and ribonuclease. Also unidentified were Hcp family proteins and a sequence described as a class SA BL PenA. 43 Hcp proteins, a family of cysteine-rich PBP, do not have a significant sequence or structural similarity to known BLs. 44 A BLASTP 34 search in the NCBI protein database 17 with the putative PenA returned as best-hit a class SC protein with only 13% coverage, meaning that it is probably not a BL and certainly not a PenA. Seven BlaR1/MecR1 proteins were also identified by the profiles but are regulatory proteins not associated with the GO term for BL.
Validation of BLASTClust thresholds to form BLs subclasses
The “BLAST score density” values applied to the curated BL sequences to form the subclasses were validated in the PDB sequence set plus NRBLD. A significant length variation was observed between the BL sequences in PDB and those in NRBLD. The sequences in PDB range from 219 to 447 amino acids, whereas in NRBLD, they range from 96 to 619 amino acids. Therefore, in addition to the previous values of “BLAST score density,” different “minimum length coverage” thresholds were chosen to cluster NRBLD sequences. Clustering results are available in Tables S5 and S6 and the thresholds in Figure 2.
Application of the similarity and coverage thresholds stipulated for clustering resulted in the separation of true BLs from other sequences such as partial domains and the regulatory proteins BlaR1/MecR1. For instance, in the case of class SA, most non-BL sequences have a larger average size (345-637 amino acids) than BLs in subclasses SA1 and SA2 (285 and 300 amino acids, respectively). One cluster contains a functionally characterized BL (LRA-5, non-BL9, Table S5). No non-BL sequence was observed for the class SC. All clusters containing non-BL sequences from class SD have one sequence, which is similar in size to BlaR1/MecR1 proteins (~585 amino acids) or partial domains (<214 amino acids). Only one of them (YP_612206, non-BL13, Table S5) is similar in size to class SD BL (274 amino acids) and its best hit in the NCBI nonredundant protein database 17 shows 43% identity with a sequence annotated as “class D BL” from Oceanicaulis alexandrii (E value of 1E−67). All non-BL sequences from class MB are partial domains (<197 amino acids). Among the 2 clusters containing non-BL sequences formed from MB1, one possesses partial domain sequences and the other has a 340-amino acid sequence from Stigmatella aurantiaca, considerably larger than BL sequences (~250 amino acids). The ME2 subclasses were not maintained after clustering of NRBLD sequences, even with higher minimum coverage thresholds (90%), not corroborating what was observed when only sequences from PDB were clustered.
Comparison of the improved profiles HMM with Pfam profiles and BL motifs
Seven Pfam 36 profiles were tested against the curated data set of BLs from PDB, showing low specificity for BL classes. The 2 Pfam profiles for SBL (PF00144.22 and PF13354.4) identified 339 and 227 enzymes out of a total of 399 SBLs, with an intersection of 220 sequences (35% and 3% CpSp, respectively). Three profiles for MBL (PF00753.25, PF12706.5, and PF16661.3) identified 98, 35, and 12 enzymes out of a total of 105 MBLs available (64%, 0%, and 0% CpSp, respectively). The other 2 MBL profiles did not identify any enzyme (PF13483.4 and PF14597.4).
Different sources were used to select 13 motifs related to the various BL classes, which were used to search among the 509 PDB sequences. About 11 specific motifs for the third classification level (SA, SC, and SD classes) and 2 motifs specific only for the second level (MBL) were used (Tables 1 and 2). The efficiency of these motifs was tested to separate BL sequences into subclasses (fourth and fifth levels).
Efficiency of serine-β-lactamase motifs.

Bush. 6
Singh et al. 45
PROSITE. 46
MACiE. 47 Target: BL class for which the motif was developed; BL: Beta-lactamase; SA: Serine-BL class SA; SC: Serine-BL class SC; SD: Serine-BL class SD.
Subclasses SA1 and SD1; **subclasses SA2 and SD2; — represents classes that have no subclass.
Efficiency of metallo-β-lactamase (MBL) motifs.

PROSITE. 46 Target: BL class for which the motif was developed; BL: Beta-lactamase; MBL: Metallo-BL; MB: Metallo-BL class MB; ME: Metallo-BL class ME; MB1: Metallo-BL subclass 1; MB2: Metallo-BL subclass MB2.
No motifs developed for MBL nor motifs for class SA are present in all the sequences allocated to their respective groups. The KxxS motif 47 was found in all class SC sequences, whereas the motif SxV 6 is present in all sequences allocated in the class SD. None of the motifs analyzed was specific to BL subclasses (MB1 or MB2, SA1 or SA2, SD1 or SD2; Tables 1 and 2).
Identification and classification of BLs in fully assembled genomes
A total of 1476 BL sequences were identified in 2774 prokaryotic genomes. SA, SC, SD, MB, and ME profiles recovered 616, 280, 366, 103, and 111 sequences, respectively. No SCD class members were found in the genomes surveyed.
After the clustering and annotation process, 123 (8.3%) sequences were considered non-BLs (Table S7). The remaining 1352 sequences (91.7%) were distributed among 12 phyla and classified according to the BL subclasses to which they belong (Table 3). All subclasses were found in the Proteobacteria phylum, excepted for MB1.2. SD1 is the most disseminated subclass among the phyla analyzed, whereas SC, SD2, and MB2 were mostly restricted to Proteobacteria. A clear difference can be observed between the phyla where BL sequences of the subclasses SA1 (Proteobacteria, Firmicutes, and Actinobacteria) and SA2 (Bacteroidetes, Cyanobacteria, and Spirochaetes) were identified. It should be noted that 49% of all analyzed strains belong to the phyla Proteobacteria. Regarding MBL subclasses, 51% (46) of MB1.1 sequences are in Firmicutes, whereas 82% (91) of ME are in Proteobacteria (Table 3). A single sequence of the MB1.2 class was identified, isolated from Sediminispirochaeta smaragdinae (WP_013255389.1), a bacteria belonging to the Spirochaetes phyla, with a specific domain of SPM-1 (cd16286).
β-lactamase sequences identified in bacterial genomes.
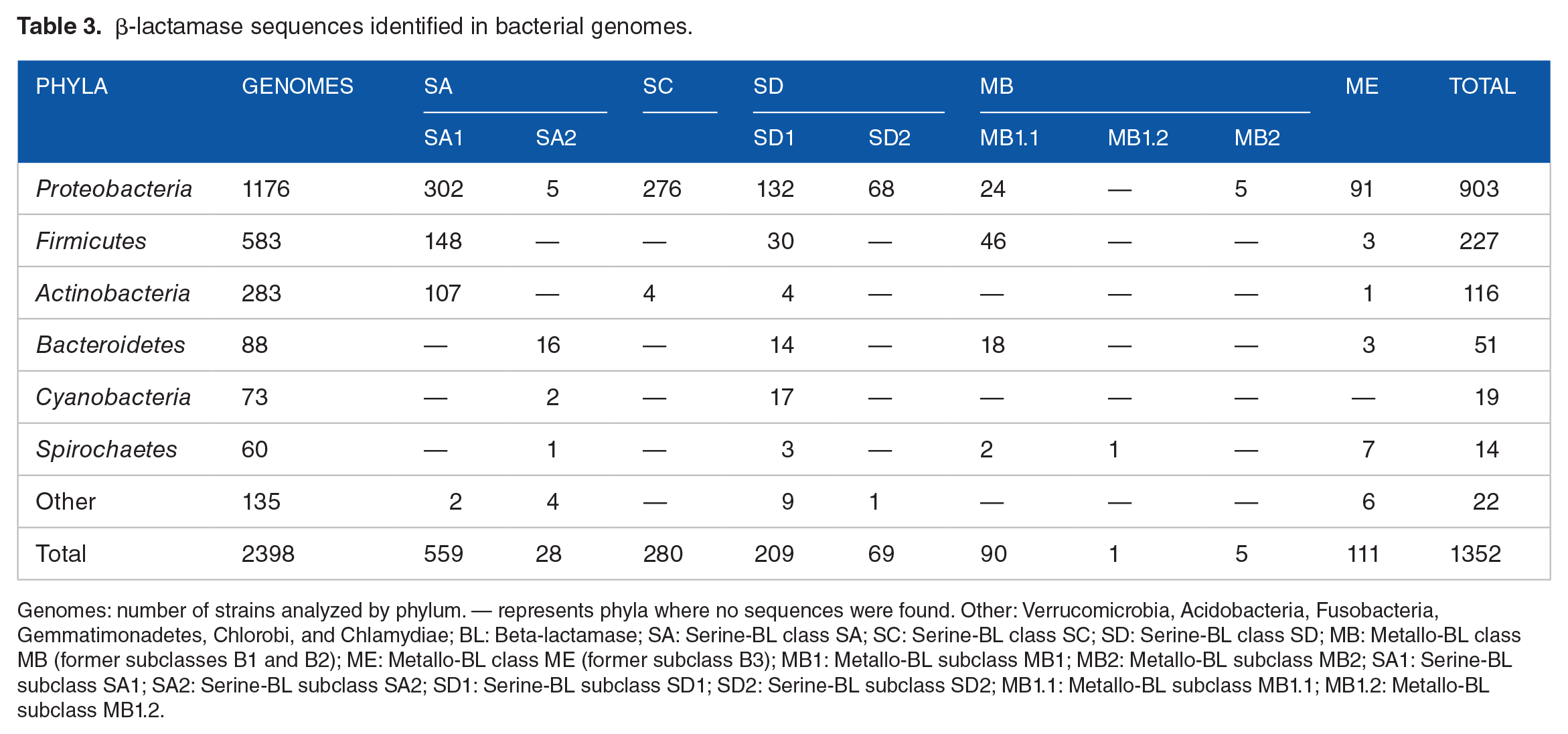
Genomes: number of strains analyzed by phylum. — represents phyla where no sequences were found. Other: Verrucomicrobia, Acidobacteria, Fusobacteria, Gemmatimonadetes, Chlorobi, and Chlamydiae; BL: Beta-lactamase; SA: Serine-BL class SA; SC: Serine-BL class SC; SD: Serine-BL class SD; MB: Metallo-BL class MB (former subclasses B1 and B2); ME: Metallo-BL class ME (former subclass B3); MB1: Metallo-BL subclass MB1; MB2: Metallo-BL subclass MB2; SA1: Serine-BL subclass SA1; SA2: Serine-BL subclass SA2; SD1: Serine-BL subclass SD1; SD2: Serine-BL subclass SD2; MB1.1: Metallo-BL subclass MB1.1; MB1.2: Metallo-BL subclass MB1.2.
A higher absolute number of BL sequences were observed in Proteobacteria. The distribution of BLs was also analyzed at the taxonomic class level for this phylum. Gammaproteobacteria class has 60% (545) of the BL sequences, divided among all BL subclasses. SD1 is the only subclass found in all classes, and the unique BL found in Epsilonproteobacteria (Table 4).
β-lactamase sequences identified in bacterial genomes from Proteobacteria phylum.

Genomes: number of strains analyzed by phylum. — represents phyla where no sequences were found BL: Beta-lactamase; SA: Serine-BL class SA; SC: Serine-BL class SC; SD: Serine-BL class SD; MB: Metallo-BL class MB (former subclasses B1 and B2); ME: Metallo-BL class ME (former subclass B3); MB1: Metallo-BL subclass MB1; MB2: Metallo-BL subclass MB2; SA1: Serine-BL subclass SA1; SA2: Serine-BL subclass SA2; SD1: Serine-BL subclass SD1; SD2: Serine-BL subclass SD2; MB1.1: Metallo-BL subclass MB1.1.
A total of 100% of the genomic sequences retrieved by the profiles (after the exclusion of non-BL sequences) were allocated to some subclass of BL. About 70% of their original annotations were designated only as BL (first level), 24% had information on the second or third level of classification or the gene name, and there were still 6% with erroneous or imprecise annotations, such as “hypothetical protein” (Figure 4).

Original (A) annotation and (B) reannotation of the 1352 sequences according to the hierarchical levels of BL classification. BL indicates β-lactamase; MBL: metallo-β-lactamase; SBL: serine-β-lactamase; MB1: Metallo-BL subclass MB1; MB2: Metallo-BL subclass MB2; SA1: Serine-BL subclass SA1; SA2: Serine-BL subclass SA2; SD1: Serine-BL subclass SD1; SD2: Serine-BL subclass SD2; ME: Metallo-BL class ME.
Discussion
Classification schemes for BLs are of utmost importance due to the diversity of these enzymes and their importance in the scenario of bacterial resistance to antibiotics.1,4,5,16 In general, the identification of new sequences is most often done by sequence comparison methods, 48 such as the BLASTP program. 34 Profiles HMM and other profile-sequence comparison methods led to a significant improvement in sensitivity over sequence comparison approaches and are already used in the identification of antibiotic resistance proteins.3,49
The workflow developed here systematizes the annotation of BLs based mainly on 2 steps: searches using profiles HMM, followed by clustering the resulting sequences. The calibrated profiles HMM can assign a sequence to a specific class. They also discriminate functionally characterized BLs from proteins with other biochemical functions that belong to the same superfamily and therefore share fold signals that make this separation difficult. 31 In addition, the calibrated profiles allowed the recognition of sequences erroneously attributed to the BL GO term (“BL activity”) in SwissProt 33 and also enable the identification of sequences imprecisely described as BL in different antibiotic resistance databases. In the clustering step, the established thresholds of similarity and coverage allowed the clearing of non-BL sequences, providing coherent BL subclasses.
Among the sequences from BL databases used to construct NRBLD, some non-BL sequences presented partial domains, suggesting assembly/sequencing artefacts or annotation errors (eg, non-BL3, Table S5). 35 Others had much larger sizes than BLs, such as the BlaR1 protein, homologous to the BLs of the class SD, but with different functions. However, 2 sequences classified as non-BL did not cluster within any BL subclass despite being similar in size to them. The LRA-5 protein, described and functionally characterized as a class SA BL, has low similarity and is a distant relative to functionally characterized BLs and their ancestors. 50 As the experimental and HMM profile data indicate that LRA-5 is a BL of class SA, we speculate that it may belong to a third subclass (SA3) considering its low similarity to other sequences. Availability of new sequences in the future will help confirm the existence of this new. The second exception (YP_612206) is 43% identical to a sequence annotated as “class D BL” of the dimorphic rods O alexandrii, although the activity of this protein has not been demonstrated. 51
It has been shown that the use of Pfam 36 profiles to identify sequences from the “Ser-BL-like superfamily” may capture unrelated sequences. 16 In our comparisons, the improved profiles HMM displayed higher specificity when compared with Pfam profiles and BL motifs from literature. Recently, individualized subgroups of the class SA have been demonstrated, such as LSBL or TEM/SHV and CARB clusters. Characteristic residues have already been attributed to each of them, but in this work we have chosen to test motifs attributed to BL classes. 1 However, subclass-specific motifs, such for SA1 and SA2, should also be tested and compared in future studies.
Some BL subclasses that were previously described based on phylogenetic criteria were identified here using sequence similarity criteria. The sequences in subclasses MB1 and MB2 correspond to the subclasses “B1” and “B2” in the work of Galleni et al. 52 The sequences in the subclass SA2 correspond to BLs isolated mainly from the group Cytophagales-Flavobacteriales-Bacteroidales.1,16,53 The OXA alleles in subclass SD2 are the same as those found in the class called “D2” by Brandt et al. 16
A new class of BLs with fused domains, the class SCD, is proposed. Two NRBLD proteins were captured by both SC and SD profiles without using the HMM bit score threshold. One of them, LRA-13, isolated from a noncultivated soil bacterium in Alaska, was confirmed experimentally as a BL, displaying a broad hydrolytic profile. 50
Subclass MB1.2 was first presented here, formed by members of the BL SPM family. It has been suggested that SPM-1 may be a structural hybrid between MB1 and MB2 subclasses. 54 SPM-1 genes were found only in isolates of Pseudomonas aeruginosa, a Proteobacteria, and its chromosomal location may have contributed to its isolation to other BL families of subclass MB1. 55 Curiously, we found that S smaragdinae, a spirochaete, carries a protein with the SPM-1 domain (WP_013255389.1). Further studies are needed to establish the evolutionary relationships between these proteins.
The distribution of BL sequences obtained from fully assembled genomes in different subclasses confirmed and complemented previous observations. For instance, the observed enrichment of BLs in Actinobacteria relative to other phyla 3 is caused mainly by members of subclass SA1. The BLs of subclass SA2, related to the Bacteroidetes phylum,1,16 were also found in other phyla. The majority presence of class SC in the Proteobacteria phylum is a consensus, and the few sequences found in Actinobacteria confirm the occasional isolation of this class. 16 The wide distribution of the subclass SD1 between the phyla analyzed confirms that class SD, more precisely the subclass SD1, has been underestimated. 16 The recently described presence of this subclass in Gram-positive bacteria has also been confirmed here. 56 The association of BL sequences from subclass SD2 to Proteobacteria may be related to the fact that they are naturally occurring intrinsic genes, most likely chromosomally located. 16 The occurrence of MB1 in Bacteroidetes and Firmicutes phyla has been associated with chromosome, whereas the Proteobacteria MB1 enzymes are mostly mobile. 57 The association of class ME with soil bacteria may be related to its distribution among different phyla, which can share the same environment. 3 New BLs of class ME have already been described in metagenomes, and the majority diverge deeply from other known enzymes of this class, 50 which reinforces the idea that class ME is widely distributed and diverse.
Acidobacteria are abundant mainly in the soil, but their cultivation is difficult. 58 Although few genomes of this phylum were available in 2016, the number of BLs identified was significant, suggesting that this is an important reservoir of BLs in nature.
The class Gammaproteobacteria includes common human pathogens such as Enterobacteriaceae and Pseudomonadaceae. 16 These pathogens are exposed continuously to evolutionary pressure exerted by antibiotics, favoring the acquisition of resistance genes, which may be related to the presence of different BL subclasses in this group. Epsilonproteobacteria, which are widely distributed bacteria including genera with pathogenic species for humans such as Helicobacter and Campylobacter, presented BLs of only the SD1 subclass. Indeed, the production of 2 major BLs (OXA-61 and OXA-184) was detected in 85% of the Campylobacter strains. 59
Conclusions
The workflow developed in this study presents better specificity when compared with available BL motifs and Pfam profiles. Application of the improved profiles HMM and sequence similarity clustering parameters resulted in a 5-level hierarchical classification, consistent with previous BL classification scheme and recent proposed subclasses based on phylogeny. We also emphasize that the number of amino acids may be an important criterion for characterizing BLs, although there may be exceptions, such as the new class of fused domain enzymes (class SCD), proposed here. The workflow presented here, which can be further improved by the addition of functional and phylogenetic data, will be of great help in studies on the prevalence, distribution, and evolution of this critical enzymatic activity.
Supplemental Material
S2_table – Supplemental material for Systematic Identification and Classification of β-Lactamases Based on Sequence Similarity Criteria: β-Lactamase Annotation
Supplemental material, S2_table for Systematic Identification and Classification of β-Lactamases Based on Sequence Similarity Criteria: β-Lactamase Annotation by Melise Chaves Silveira, Rangeline Azevedo da Silva, Fábio Faria da Mota, Marcos Catanho, Rodrigo Jardim, Ana Carolina R Guimarães and Antonio B de Miranda in Evolutionary Bioinformatics
Supplemental Material
S4_table – Supplemental material for Systematic Identification and Classification of β-Lactamases Based on Sequence Similarity Criteria: β-Lactamase Annotation
Supplemental material, S4_table for Systematic Identification and Classification of β-Lactamases Based on Sequence Similarity Criteria: β-Lactamase Annotation by Melise Chaves Silveira, Rangeline Azevedo da Silva, Fábio Faria da Mota, Marcos Catanho, Rodrigo Jardim, Ana Carolina R Guimarães and Antonio B de Miranda in Evolutionary Bioinformatics
Supplemental Material
S6_nova – Supplemental material for Systematic Identification and Classification of β-Lactamases Based on Sequence Similarity Criteria: β-Lactamase Annotation
Supplemental material, S6_nova for Systematic Identification and Classification of β-Lactamases Based on Sequence Similarity Criteria: β-Lactamase Annotation by Melise Chaves Silveira, Rangeline Azevedo da Silva, Fábio Faria da Mota, Marcos Catanho, Rodrigo Jardim, Ana Carolina R Guimarães and Antonio B de Miranda in Evolutionary Bioinformatics
Supplemental Material
uomf_july_23_2018_s1_table – Supplemental material for Systematic Identification and Classification of β-Lactamases Based on Sequence Similarity Criteria: β-Lactamase Annotation
Supplemental material, uomf_july_23_2018_s1_table for Systematic Identification and Classification of β-Lactamases Based on Sequence Similarity Criteria: β-Lactamase Annotation by Melise Chaves Silveira, Rangeline Azevedo da Silva, Fábio Faria da Mota, Marcos Catanho, Rodrigo Jardim, Ana Carolina R Guimarães and Antonio B de Miranda in Evolutionary Bioinformatics
Supplemental Material
uomf_july_23_2018_s3_table – Supplemental material for Systematic Identification and Classification of β-Lactamases Based on Sequence Similarity Criteria: β-Lactamase Annotation
Supplemental material, uomf_july_23_2018_s3_table for Systematic Identification and Classification of β-Lactamases Based on Sequence Similarity Criteria: β-Lactamase Annotation by Melise Chaves Silveira, Rangeline Azevedo da Silva, Fábio Faria da Mota, Marcos Catanho, Rodrigo Jardim, Ana Carolina R Guimarães and Antonio B de Miranda in Evolutionary Bioinformatics
Supplemental Material
uomf_july_23_2018_s5_table – Supplemental material for Systematic Identification and Classification of β-Lactamases Based on Sequence Similarity Criteria: β-Lactamase Annotation
Supplemental material, uomf_july_23_2018_s5_table for Systematic Identification and Classification of β-Lactamases Based on Sequence Similarity Criteria: β-Lactamase Annotation by Melise Chaves Silveira, Rangeline Azevedo da Silva, Fábio Faria da Mota, Marcos Catanho, Rodrigo Jardim, Ana Carolina R Guimarães and Antonio B de Miranda in Evolutionary Bioinformatics
Supplemental Material
uomf_july_23_2018_s6_table – Supplemental material for Systematic Identification and Classification of β-Lactamases Based on Sequence Similarity Criteria: β-Lactamase Annotation
Supplemental material, uomf_july_23_2018_s6_table for Systematic Identification and Classification of β-Lactamases Based on Sequence Similarity Criteria: β-Lactamase Annotation by Melise Chaves Silveira, Rangeline Azevedo da Silva, Fábio Faria da Mota, Marcos Catanho, Rodrigo Jardim, Ana Carolina R Guimarães and Antonio B de Miranda in Evolutionary Bioinformatics
Supplemental Material
uomf_july_23_2018_s7_table – Supplemental material for Systematic Identification and Classification of β-Lactamases Based on Sequence Similarity Criteria: β-Lactamase Annotation
Supplemental material, uomf_july_23_2018_s7_table for Systematic Identification and Classification of β-Lactamases Based on Sequence Similarity Criteria: β-Lactamase Annotation by Melise Chaves Silveira, Rangeline Azevedo da Silva, Fábio Faria da Mota, Marcos Catanho, Rodrigo Jardim, Ana Carolina R Guimarães and Antonio B de Miranda in Evolutionary Bioinformatics
Footnotes
Acknowledgements
The authors would like to thank Dr Reema Singh and Dr Harpreet Singh for providing the DLact database sequences and Dr Alex Herbert for giving us the script to remove alternative positions of the atoms. MCS recognizes CAPES (Coordination for the Improvement of Higher Education Personnel, Brazil) for supporting her with a scholarship during her DSc program.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
MCS and ABM conceived of the manuscript outline; MCS, FFM, MC, RJ, ACRG, and ABM jointly developed the methodology; MCS, RAS, FFM, MC, and ABM wrote, edited and revised the manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.