
Abstract
Amyloids are protein fibrils with a highly ordered spatial structure called cross-β. To date, amyloids were shown to be implicated in a wide range of biological processes, both pathogenic and functional. In bacteria, functional amyloids are involved in forming biofilms, storing toxins, overcoming the surface tension, and other functions. Rhizobiales represent an economically important group of Alphaproteobacteria, various species of which are not only capable of fixing nitrogen in the symbiosis with leguminous plants but also act as the causative agents of infectious diseases in animals and plants. Here, we implemented bioinformatic screening for potentially amyloidogenic proteins in the proteomes of more than 80 species belonging to the order Rhizobiales. Using SARP (Sequence Analysis based on the Ranking of Probabilities) and Waltz bioinformatic algorithms, we identified the biological processes, where potentially amyloidogenic proteins are overrepresented. We detected protein domains and regions associated with amyloidogenic sequences in the proteomes of various Rhizobiales species. We demonstrated that amyloidogenic regions tend to occur in the membrane or extracellular proteins, many of which are involved in pathogenesis-related processes, including adhesion, assembly of flagellum, and transport of siderophores and lipopolysaccharides, and contain domains typical of the virulence factors (hemolysin, RTX, YadA, LptD); some of them (rhizobiocins, LptD) are also related to symbiosis.
Keywords
Introduction
The term “amyloid” refers to fibrillary protein aggregates possessing highly ordered spatial structure called “cross-β.” 1 Protein monomers comprising amyloid fibrils form repetitive intermolecular β-sheets stabilized by numerous hydrogen bonds 2 that result in a specific X-ray diffraction pattern. 3 This makes amyloids highly resistant to different chemicals, enzymes, and physical factors. 4 The ability of a protein to adopt amyloid state is determined by the presence of so-called amyloidogenic regions (ARs)5,6 in its amino acid sequence, which act as inducers of amyloid formation. 7 There are at least 2 types of ARs identified to date. Type I ARs are formed by the compositionally biased regions rich in glutamine (Q) or asparagine (N) and are crucial for amyloid formation in different organisms ranging from fungi 8 to humans. 9 Type I ARs are efficiently predicted by LPS (Lower Probability Subsequences) 10 and SARP (Sequence Analysis based on the Ranking of Probabilities) 11 bioinformatics algorithms. Type II ARs are formed by different hydrophobic amino acids (I, L, V, F, W, Y), 12 and in contrast to Type I ARs where amino acid composition is more important, 13 the position of each particular residue is crucial for the formation of Type II ARs. 14 Type II ARs are predicted by various bioinformatics algorithms, 12 one of the most efficient of which is Waltz. 14 Bioinformatic prediction is used to select the promising candidates for further experimental confirmation of their potential amyloid properties.15,16
Amyloids are well known as the lethal pathogens associated with dozens of incurable diseases of humans and animals.1,17 The term “amyloid” had been initially introduced by Rudolf Virchow 18 for pathological iodine-positive deposits in human tissues, whereas the protein nature of such deposits was revealed later. 1 At least 30 proteins were reported to form pathological amyloids in human tissues. 19 The progress in the investigation of amyloids has led to reinterpretation of their biological roles. Since 2000, about 25 proteins of different organisms, from archaea and bacteria to humans, have been shown to adopt functional amyloid state under native conditions. 20 The greatest diversity of functional amyloids was found in prokaryotes. To date, only 2 groups of proteins were shown to form such amyloids under native conditions in archaea.21,22 At the same time, the number of groups of functional amyloids in bacteria is about 10, and the spectrum of their functions involves, but is not limited to, toxin storage, 23 biofilm formation, 24 and development of aerial hyphae 25 and spores. 26
Proteobacteria represents the largest phylum in the domain Bacteria comprising about one-third of the known bacterial species and consisting of 5 classes: Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria, Deltaproteobacteria, and Epsilonproteobacteria. To date, amyloid-forming proteins were identified in different pathogenic proteobacteria species belonging to the class Gammaproteobacteria (Escherichia coli, Salmonella enteritidis, Pseudomonas aeruginosa, Klebsiella pneumonia, etc). 20
Bioinformatic prediction is a powerful tool to reveal prominent candidates for further experimental verification of their amyloid properties.27–29 In this study, we focused on predicting the potentially amyloidogenic proteins in the species of the order Rhizobiales, which belongs to the class Alphaproteobacteria. Rhizobiales represent a uniquely diverse group of bacteria, embracing not only species capable of nitrogen fixation in the symbiosis with the leguminous plants but also dangerous pathogens of animals and plants. 30 The investigation of potentially amyloidogenic proteins in Rhizobiales has not been carried out before and is of great interest because of the unusual ecological and functional diversity of these bacteria. We implemented a large-scale analysis of potentially amyloidogenic proteins in the proteomes of more than 80 species of Rhizobiales by means of SARP and Waltz bioinformatics algorithms. We identified biological processes, where the potentially amyloidogenic proteins are overrepresented, analyzed their subcellular localization, and identified the protein domains and regions associated with the presence of ARs. Our data reveal that potentially amyloidogenic proteins of Rhizobiales tend to be functionally associated with the symbiotic and pathogenic properties of these bacteria.
Materials and Methods
Datasets
Proteomes of 87 strains of different species of the order Rhizobiales (Table S1) were downloaded from the UniProt database (http://uniprot.org/proteomes/). Their systematic positions were based on the UniProt Taxonomy database (http://uniprot.org/taxonomy/). All annotations of proteins including Gene Ontology (GO, http://www.geneontology.org/) terms annotation and structural features of proteins were downloaded from the UniProt database (http://uniprot.org/). To obtain data from UniProt, we used the Proteins REST API (http://www.ebi.ac.uk/proteins/api/doc). 31
Prediction of compositionally biased regions
Compositionally biased regions enriched with glutamine and asparagine were predicted by SARP program. 11 The probability threshold was 10–8. The minimal length of the regions was 15 amino acids. If protein contained at least 1 QN-rich region, it was considered potentially amyloidogenic. The coverage of compositionally biased regions was calculated as follows: the sum of lengths of all QN-rich regions was divided by the sum of lengths of all proteins for each proteome.
Prediction of ARs
Short ARs were predicted by Waltz 14 with the best overall selectivity threshold and pH 7.0. Waltz requires the protein sequence to be no longer than 10 000 amino acids and not contain uncanonical amino acids, so such proteins were excluded from analysis. Protein was considered potentially amyloidogenic if it harbored at least 1 region predicted by Waltz and longer than 9 amino acids. Coverage of Waltz-predicted regions was calculated the same as for SARP-predicted QN-rich regions.
GO enrichment
We used a topGO R package 32 to perform GO enrichment test. The GO terms annotation was obtained from UniProt database. Proteins predicted to be potentially amyloidogenic by Waltz or SAPR were tested against the list of all proteins for each proteome. We selected only GO terms with P-value lower than .01 and with at least 5 proteins assigned to a given term. The GO terms were ordered by the number of proteomes where they were enriched with potentially amyloidogenic proteins. Heatmaps were drawn with heatmap.2 function from gplots package. Phylogenetic tree was used to clusterize proteomes.
Coverage of protein structural features by ARs
All structural elements of proteins were obtained from UniProt database (http://www.uniprot.org/). Protein regions not assigned to any structural element were marked as unannotated. For each type of structural elements, the sum of lengths of overlapping regions between elements of given type and ARs predicted by Waltz or SAPR was divided by the sum of lengths of those elements. Types of elements were ordered by the average value of coverage across all proteomes. Heatmaps were drawn the same way as for GO terms enrichment.
Results
Analyzing the distribution of potentially ARs in the proteomes of Rhizobiales
To estimate the abundance of potentially amyloidogenic proteins in the Rhizobiales bacteria, we predicted ARs in the proteomes of 86 species of this order with 2 different algorithms, Waltz and SARP (Figure 1). Waltz predicts very short ARs with a median length of approximately 6 residues in many proteins, and the frequency of the regions of given length decreases drastically with an increase in length, 14 so to decrease the amount of false-positive predictions, we analyzed only the regions longer than 9 amino acids. The data obtained show the Rhizobiales species to be very different by the amount of proteins with ARs predicted with Waltz and QN-rich regions predicted with SARP (Figure 1). The percentage of proteins containing such regions predicted with Waltz varied from 11.7% to 22.3% of the total number of proteins in the proteome, while the same for SARP varied from 0.2% to 1.4% (Figure 1). Mostly, no more than 10% of the amino acid sequences of proteins were covered by the ARs predicted with Waltz (Figure S1). In contrast, the QN-rich regions predicted with SARP covered from 20% to 100% of the sequences of proteins (Figure S2). The fractions of proteins containing the ARs predicted by Waltz were relatively similar even among evolutionary distant species (Figure 1), whereas the fractions of QN-rich proteins varied more significantly and were markedly different even for the species of the same genus (Figure 1). Nonetheless, several genera, like Nitratireductor or Devosia, are characterized by very similar fractions of QN-rich proteins among the species comprising them (Figure 1).

Phylogenetic tree indicating the distribution of potentially amyloidogenic proteins predicted with Waltz and SARP (Sequence Analysis based on the Ranking of Probabilities) in the proteomes of Rhizobiales. The percentage of proteins harboring potentially amyloidogenic regions predicted with Waltz, and QN-rich regions found with SARP, to the total number of proteins in the proteome (light gray) and the percentage of the length of these regions to the total length of all proteins in the proteome (dark gray) are shown.
Subcellular localization of potentially amyloidogenic proteins
Localization of proteins is a key feature indicating their involvement in the specific groups of biological processes. To assess whether the potentially amyloidogenic proteins have preferred cellular localization, we performed a GO terms enrichment test and selected subcellular localization GO terms for which the potentially amyloidogenic proteins predicted either with Waltz or with SARP were overrepresented with a probability lower than .01. We found that QN-rich proteins predicted by SARP were overrepresented among the proteins of the extracellular region, cell outer membrane, as well as structures involved in the cell motility, mostly flagellum (Figure 2). Most of the potentially amyloidogenic proteins harboring Waltz-predicted regions were associated with the plasma membrane and several high-molecular-weight protein complexes (respiratory chain and cytochromes) (Figure S3). Thus, the potentially amyloidogenic proteins of Rhizobiales have mostly plasma membrane, outer membrane, or extracellular localization.
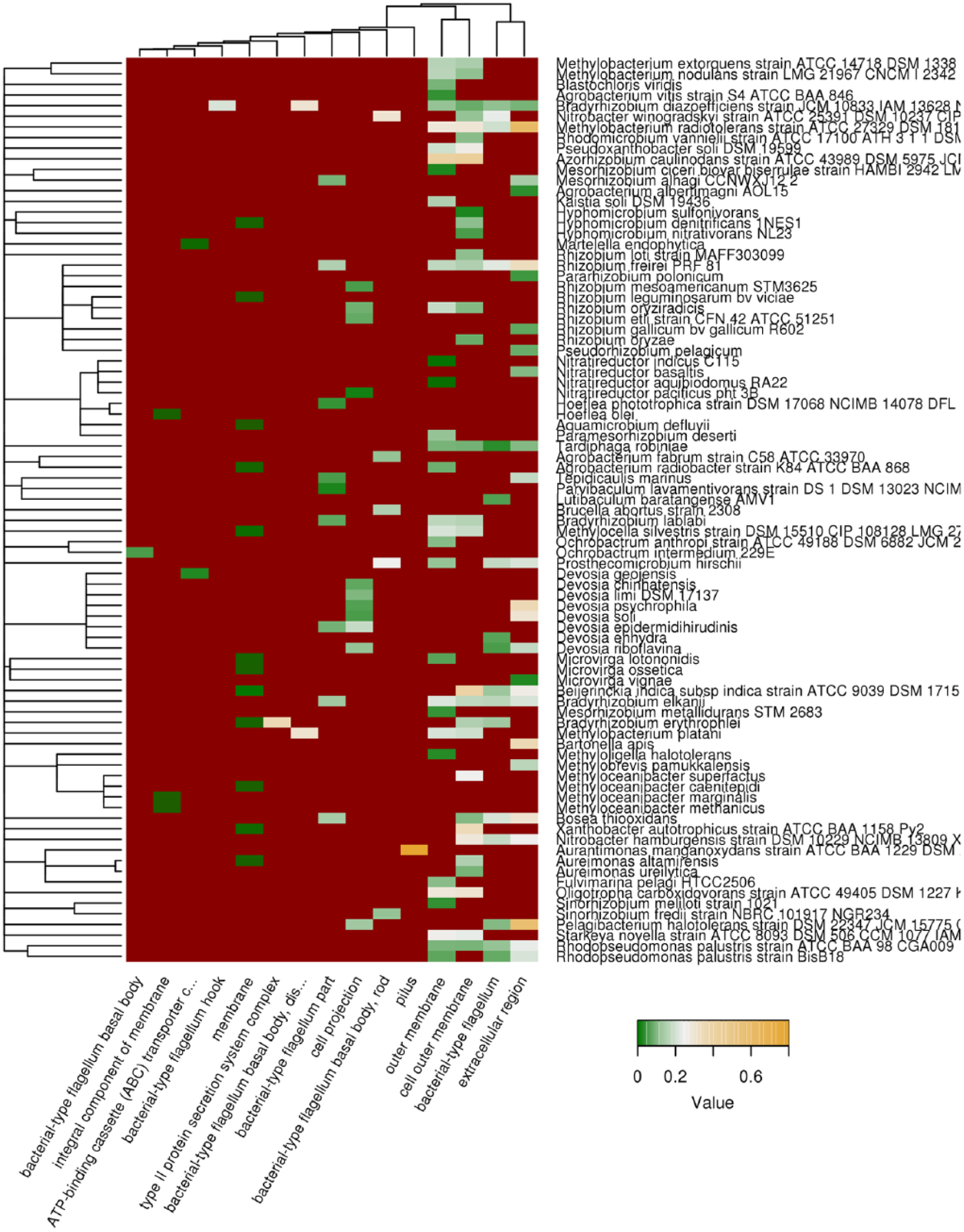
Subcellular localization of QN-rich potentially amyloidogenic proteins predicted with SARP (Sequence Analysis based on the Ranking of Probabilities). Top 15 cellular localizations according to the Gene Ontology (GO) database that are most abundant among QN-rich proteins predicted with SARP are shown. Color of cells denotes fraction of QN-rich proteins to all proteins in given category. Dark red denotes that the category is not overrepresented. Tree of species corresponds to their phylogeny.
Biological processes in which potentially amyloidogenic proteins of Rhizobiales are implicated
Proteins with the same localizations can take part in various biological processes. Therefore, we obtained the list of biological processes according to the GO database, for which proteins with ARs were overrepresented. The results obtained show that the proteins bearing the regions predicted with Waltz were abundant among different proteins involved in the transport through plasma membrane, adenosine triphosphate (ATP) synthesis coupled electron transport, aerobic electron transport chain, as well as secretion (Figure 3), thus confirming their predominant plasma membrane localization.
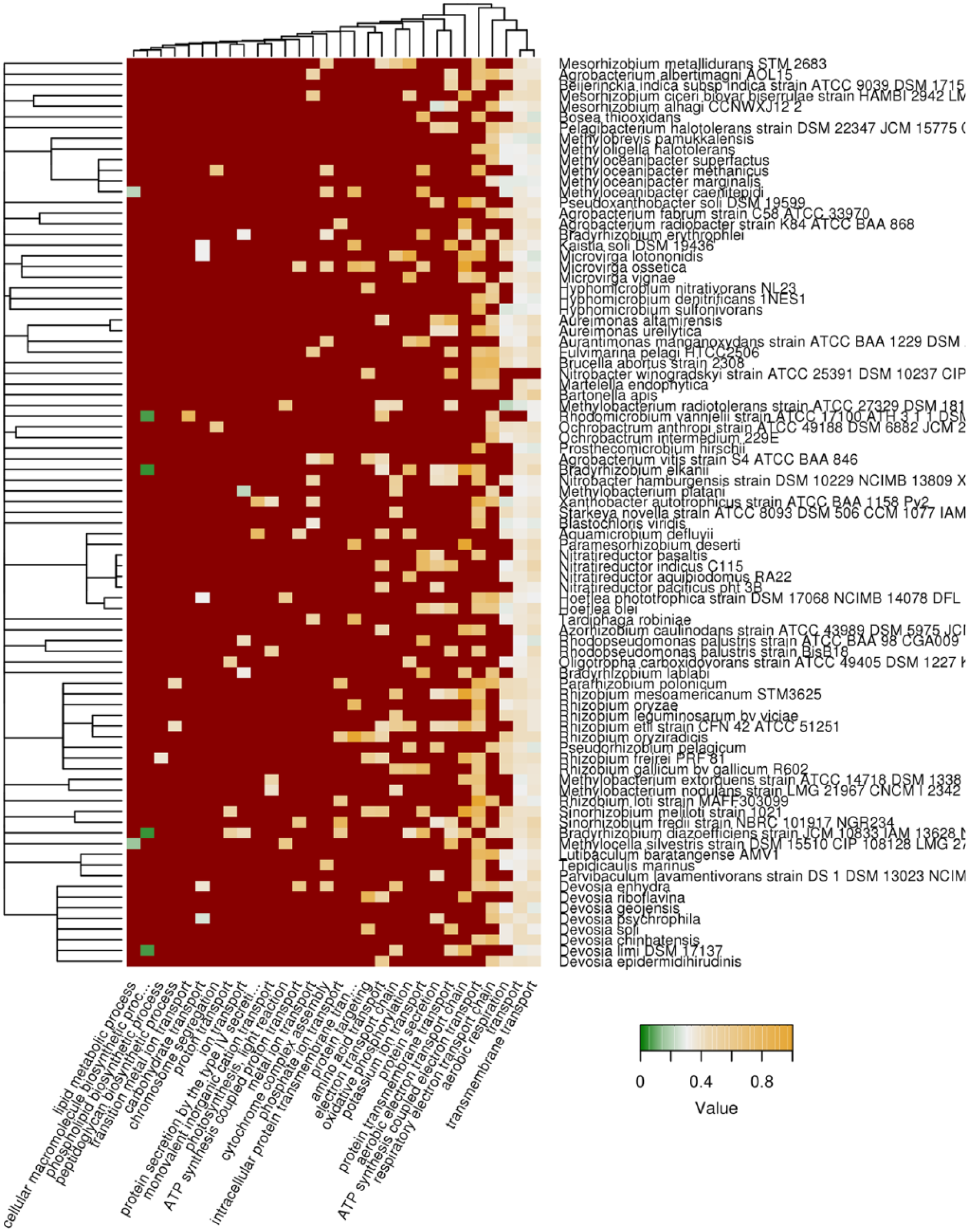
Biological processes associated with potentially amyloidogenic proteins predicted with Waltz. Top 30 biological processes according to the Gene Ontology (GO) database, most abundant with proteins with potentially amyloidogenic regions predicted by Waltz are shown. Color of cells denotes fraction of potentially amyloidogenic proteins to all proteins in given category. Dark red denotes that the category is not overrepresented. Tree of species corresponds to their phylogeny.
At the same time, QN-rich proteins predicted by SARP were involved in a broader spectrum of biological processes, including transmembrane transport, transport of siderophores, biosynthesis of polysaccharides, cytokinesis, protein secretion, and stress response (Figure 4). A significant fraction of biological processes with abundance of QN-rich proteins was associated with motility and flagellum assembly, with QN-rich proteins abundant in the bacterial-type flagellum-dependent cell motility process in about half of the species (Figure 4). Interestingly, Aurantimonas manganoxydans was the only species, for which cell adhesion proteins, components of bacterial pilus, were enriched with QN-rich regions (Figures 2 and 4). Seven other Rhizobiales species had QN-rich proteins abundant in the pathogenesis processes.

Biological processes associated with QN-rich proteins predicted by SARP (Sequence Analysis based on the Ranking of Probabilities). Top 30 biological processes according to the Gene Ontology (GO) database, most abundant with proteins with QN-rich regions predicted with SARP are shown. Color of cells denotes fraction of QN-rich proteins to all proteins in given category. Dark red denotes that the category is not overrepresented. Tree of species corresponds to their phylogeny.
It should be noted that some known functional bacterial amyloids take part in the processes of host invasion and pathogenesis.33,34 So, we obtained the list of all proteins linked with the “pathogenesis” GO term (GO:0009405) and bearing QN-rich regions predicted by SARP (Table 1). The analysis of these proteins in UniProt database (http://www.uniprot.org/) showed they had YadA, RTX, or hemolysin-type domains known to be strongly associated with the bacterial pathogenesis.
QN-rich potentially amyloidogenic proteins associated with pathogenicity of Rhizobiales.

Protein domains in which ARs are overrepresented
Next, we performed more comprehensive search for protein domains associated with the ARs in the proteomes of Rhizobiales. Such regions could be located inside the functional domains or reside in unannotated nonfunctional regions. So, we calculated the percentage of the lengths of different domains covered by ARs predicted with Waltz or SARP. Most of regions predicted with both, Waltz and SARP, are located inside unannotated unstructured regions; the rest of QN-rich regions tend to be located in β-chains, and regions predicted with Waltz are mostly located in helical transmembrane domains (Figures 5 and 6). We revealed that the density of regions predicted with Waltz was highest in the cytochrome-C-oxidase–like domains as well as different transporter domains (Figure 5) that corresponds to the data that the cytochrome complexes and transmembrane transporters are enriched with the Waltz-predicted amyloidogenic proteins (Figure S3 and Figure 3).

Protein domains associated with amyloidogenic regions (ARs) predicted by Waltz. Protein domains and structural features most enriched with amyloidogenic region predicted with Waltz are shown. Color of cells denotes fraction of the length of potentially amyloidogenic regions harbored by the feature to the total length of the given feature in all proteins. Dark red denotes the absence of proteins with such domain in the proteome of the given bacterial species. Tree of species corresponds to their phylogeny.
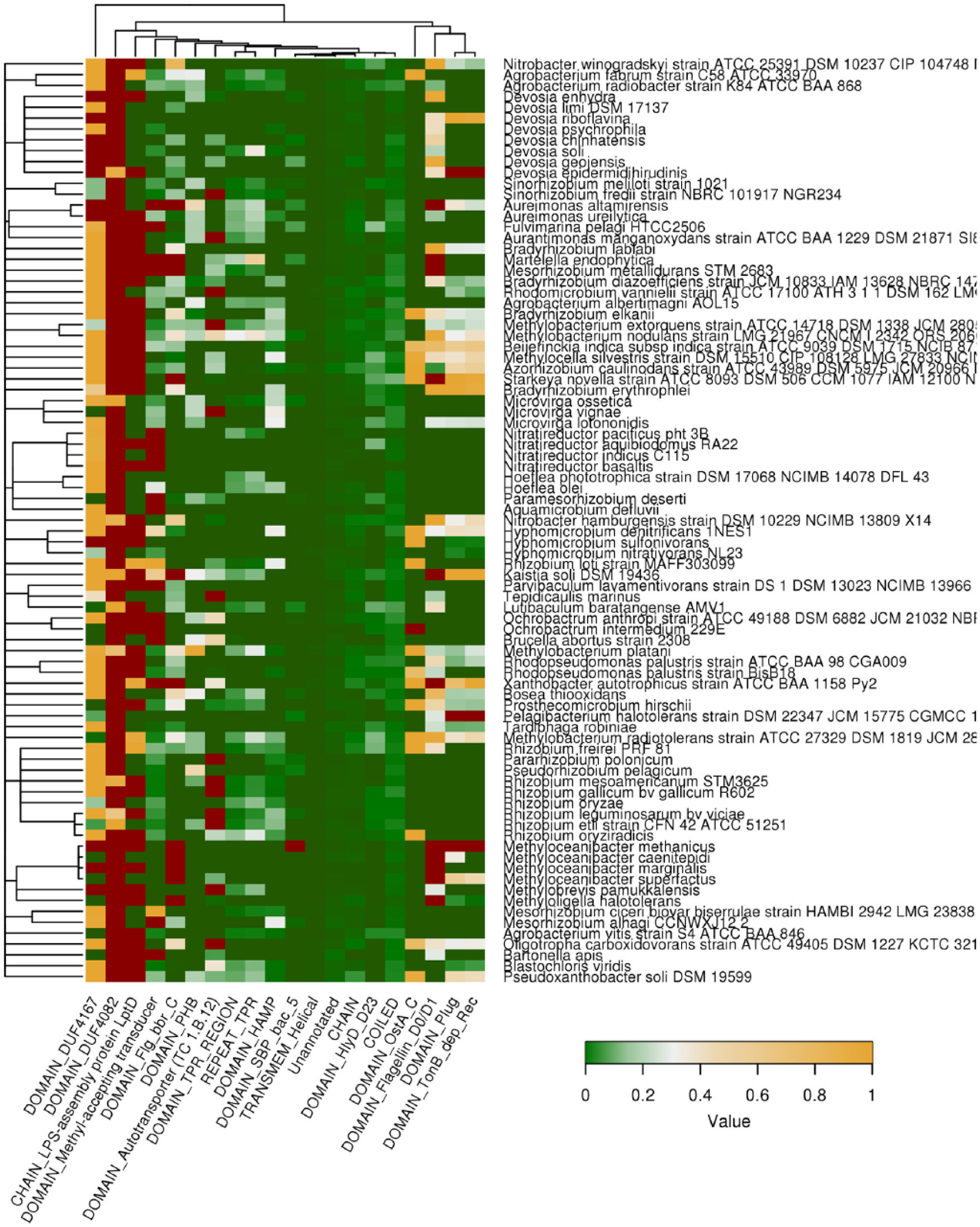
Protein domains associated with QN-rich compositionally biased regions (CBRs) predicted by SARP (Sequence Analysis based on the Ranking of Probabilities). Protein domains and structural features most enriched with QN-rich region predicted by SARP are shown. Color of cells denotes fraction of the length of QN-rich regions harbored by the feature to the total length of the given feature in all proteins. Dark red denotes the absence of proteins with such domain in the proteome of the given bacterial species. Tree of species corresponds to their phylogeny.
The analysis of the association between QN-rich regions predicted with SARP and protein domains revealed that highest density of QN-rich regions was in several domains of unknown functions (DUF4167 and DUF4082), flagellin (Flg) and hemolysin-type (HlyD) domains, as well as domains involved in the biosynthesis of lipopolysaccharides (LptD) and secretion (Figure 6). Therefore, ARs are associated with the same functional groups of proteins (transporters, flagellar proteins, and pathogenesis-related) that were revealed in the analysis of biological processes (Figures 3 and 4). The presence of these domains in the bacterial species, as well as their coverage with ARs, is shown in Figure S4 for regions predicted by Waltz and in Figure S5 for QN-rich regions. The most part of domains associated with ARs predicted with Waltz or SARP are conservative in Proteobacteria, with COX1 and COX3 domains conservative in all living organisms. Taking together, ARs tend to co-occur within the domains of proteins with specific molecular functions related to transport and pathogenesis.
Discussion
Rhizobiales represents a unique group of microbes comprising both highly specialized symbionts and hazardous pathogens of multicellular organisms. 35 The amyloids of bacteria are known to be involved in various functions, the most studied of which is biofilm formation which is important for the virulence and host-pathogen interactions. 36 The analysis carried out in this study demonstrated that the potentially amyloidogenic proteins of Rhizobiales predicted with Waltz had plasma membrane localization (Figure S3), whereas a significant part of proteins predicted with SARP were located within the outer membrane or had the extracellular localization (Figure 2), which are typical of the virulence proteins. Interestingly, the majority of QN-rich proteins in eukaryotes have cytoplasmic localization, 10 whereas bacterial QN-rich proteins are mainly membrane or secreted proteins (Figure 2). Given this evidence, we hypothesized that amyloids of Rhizobiales could be involved in the symbiosis, which, similar to pathogenesis, is mediated by different extracellular proteins. 37
The fractions of amyloidogenic proteins predicted with SARP and Waltz significantly varied in different species and were not associated with their pathogenic or symbiotic life cycle features (Figure 1). At the same time, we revealed both—biological processes (Figures 3 and 4) and protein domains (Figures 5 and 6) related to potentially amyloidogenic proteins of Rhizobiales—and determined a strong association between the ARs and pathogenesis of bacteria. For instance, a significant number of potentially amyloidogenic proteins of Rhizobiales predicted with Waltz and SARP act as the transmembrane transporters and channels (Figures 3 and 4). These transmembrane proteins include porins; 2 of them—OmpA 38 and OmpC 34 —were shown to have amyloid properties at least in vitro and represent important virulence factors of E coli. 39 The second group of potentially amyloidogenic QN-rich proteins of Rhizobiales was flagellar proteins (Figure 4) containing different Flg-like domains (Figure 6). In addition to the locomotion function, flagellum is known to play an important role in the adhesion and virulence of different bacteria, 40 including Brucella belonging to the order Rhizobiales. 41 We also found that QN-rich proteins of most of the Rhizobiales species were involved in cell adhesion (Figure 4), which plays a crucial role in the pathogenesis of bacteria. 42 Transport of siderophores, low-weight iron-chelating molecules, which was found to be associated with QN-rich proteins predicted by SARP (Figure 4), is an important factor in the development of various bacterial infections. 43 The LptD-like domain predicted by SARP as potentially amyloidogenic is typical of the outer membrane proteins responsible for lipopolysaccharide (LPS) transport that is important for pathogenesis 44 and symbiosis. 45 Finally, the analysis of potentially amyloidogenic QN-rich proteins detected by SARP and associated with pathogenesis (Table 1) demonstrated that they comprise at least 3 functionally related families of protein domains: YadA-like, RTX toxin and hemolysin-like. YadA is an adhesion protein initially discovered in Yersinia, virulence factor which binds to various substrates and acts as invasin. 46 There are 2 types of YadA domains—YadA stalk and YadA anchor—which were found in 16 different species of Rhizobiales. Only YadA stalk, which is considered to take part in the polymerization of YadA-like proteins, was associated with QN-rich regions (Figure S5). 47 RTX toxins are the pore-forming endotoxins accounting for the major virulence factors of different Proteobacteria species. 48 Hemolysin-like domains are associated with various proteins, including metalloproteases, and different toxins, including rhizobiocins, the calcium-dependent bacteriocins produced by Rhizobium species to protect from other bacterial species that affect the effectiveness of symbiosis. 49
To sum up, we may conclude that the potentially amyloidogenic QN-rich proteins of Rhizobiales exhibit a strong association with pathogenesis and virulence of this group of bacteria. They have mainly a membrane or extracellular localization and are functionally involved in various processes associated with virulence and infection (cell adhesion, siderophore and LPS transport, assembly of flagellum) and contain domains typical of the virulence factors (hemolysin, RTX, YadA, LptD); some of them (rhizobiocins, LptD) are also related to symbiosis. Overall, our data support the hypothesis according to which amyloid formation by various proteins could play a crucial role in the virulence of bacteria. Further experimental verification of amyloid properties of the virulence proteins will highlight the role of amyloid state in bacterial pathogenesis.
Supplemental Material
Supplementary – Supplemental material for Exploring Proteins Containing Amyloidogenic Regions in the Proteomes of Bacteria of the Order Rhizobiales
Supplemental material, Supplementary for Exploring Proteins Containing Amyloidogenic Regions in the Proteomes of Bacteria of the Order Rhizobiales by Kirill S Antonets, Sergey F Kliver and Anton A Nizhnikov in Evolutionary Bioinformatics
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was financially supported by the Russian Science Foundation (grant No 17-16-01100).
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
KSA and AAN conceived and designed the experiments; KSA and AAN analyzed the data; KSA, SFK, and AAN wrote the first draft of the manuscript; KSA, SFK, and AAN agree with manuscript results and conclusions; KSA and AAN jointly developed the structure and arguments for the paper; KSA, SFK, and AAN made critical revisions and approved the final version. All authors reviewed and approved the final manuscript.
Disclosures and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including, but not limited to, the following: authorship and contributorship, conflicts of interest, privacy and confidentiality, and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.