
Abstract
Purpose
To report and document a case of torpedo maculopathy found in a patient affected by keratoconus.
Case report: An healthy 16-year-old male patient, affected by keratoconus in both eyes, was referred to the cornea service of our hospital for a follow-up visit.
During the dilated fundus examination of the left eye, an oval, well-demarcated, hypopigmented lesion was observed in the juxtafoveal temporal region, pointing towards the center of the macula. Multimodal imaging of the lesion was performed, and the diagnosis of Torpedo Maculopathy was established based on the clinical picture.
Conclusion
This is the first case of torpedo maculopathy described in a patient affected by keratoconus. This association may be merely fortuitous or the result of developmental abnormalities affecting both corneal and retinal structures.
Keywords
Introduction
Torpedo maculopathy (TM) is a rare unilateral developmental anomaly of the retinal pigment epithelium (RPE)/choriocapillaris complex, with a prevalence of 2/100,000 1 to 17/55,334. 2 It manifests as variable defects in the outer layers of the retina and four types have been described.3–5
In 1992, Roseman and Gass 6 described the features of a “hypopigmented nevus of the retinal pigment epithelium,” later termed torpedo maculopathy by Daily MJ. 7 This condition is characterized by an asymptomatic oval torpedo-shaped defect in the RPE, specifically located in the temporal macular region, with a pointed tip directed toward the foveola. While bearing resemblance to solitary congenital hypertrophy of the RPE (CHRPE), it differs in terms of its nonrandom macular location and torpedo-like shape. 8 The exact pathogenesis of this entity remains unknown and speculative. Shields et al. 8 hypothesized, based on Streeten's research on fetal RPE development, that TM may indicate a persistent anomaly in the developmental process of the RPE within the fetal temporal bulge. Previous reported cases have not demonstrated any systemic associations.
Keratoconus (KC) is an uncommon bilateral ectatic disorder, with a worldwide prevalence of approximately 1/2,000. 9 It is characterized by central or paracentral progressive thinning and steepening of the cornea, leading to irregular astigmatism. The pathophysiology of KC likely involves genetic, environmental (e.g., chronic eye rubbing, atopy of the eye, and contact lens wear), and biochemical factors (e.g., upregulation of degradative enzymes). According to an extensive body of literature,9–11 an initial event, which is yet to be determined and potentially regulated by genetic factors, initiates the failure of the “locking mechanism” established during early childhood, resulting in the destabilization of collagen fibrils in the corneal stroma. Subsequent biomechanical and biochemical processes contribute to the occurrence of lamellar or fibrillar slippage in the cornea, leading to the typical KC changes. 10
To the best of our knowledge, no case reports have been found associating TM and KC. Therefore, we present the first reported case of TM in a patient affected by KC. TM is a poorly studied pathology, while KC is an etiologically heterogeneous disorder with not entirely known genetic causes and pathogenesis. Reporting this unusual association can provide valuable insights into these two rare diseases.
Case report
An otherwise healthy 16-year-old male patient, who had undergone collagen cross-linking for keratoconus in both eyes at the age of 13, presented for a follow-up visit at the cornea service of our Ophthalmology Department (Fondazione Policlinico Universitario A. Gemelli IRCCS, Rome, Italy). The best corrected visual acuity was 20/25 and 20/20, with a refractive error of +0,50=-5,00(60°) and +0,25=-3,50(95°) in the right and left eye, respectively (Figure 1A). The retinography of the left eye revealed an oval, well-demarcated, hypopigmented lesion in the juxtafoveal temporal region, pointing towards the center of the macula, encircled by a pigmented ring and with a slight tail to its temporal edge (Figure 1B).
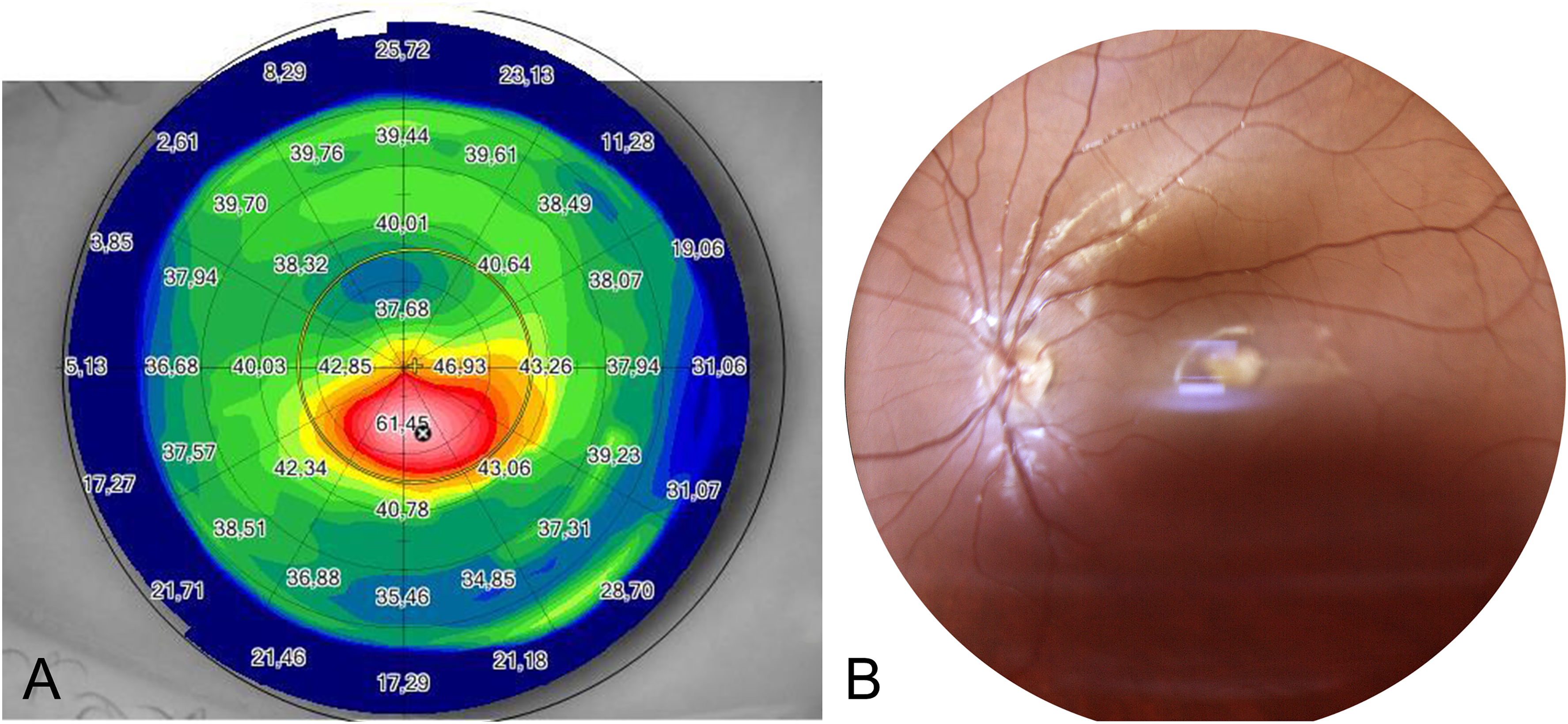
(A) The tangential anterior map of the left eye, obtained using a combined rotating Scheimpflug camera and Placido-disk corneal topographer (Sirius, CSO, Italy), reveals an apex keratometric Reading of 61.45 diopters and eccentric corneal steepening. (B) Additionally, the true-color fundus photograph captured by a confocal light-emitting diode-based retinal imaging system (Eidon, Centervue, Padova, Italy) demonstrates the presence of a torpedo-like lesion in the temporal juxtafoveal area. It should be noted that the defocus blur in the image is due to the presence of keratoconus.
The fundus autofluorescence showed a homogenously hypoautofluorescent area, surrounded by an incomplete hyperautofluorescent ring (Figure 2B).
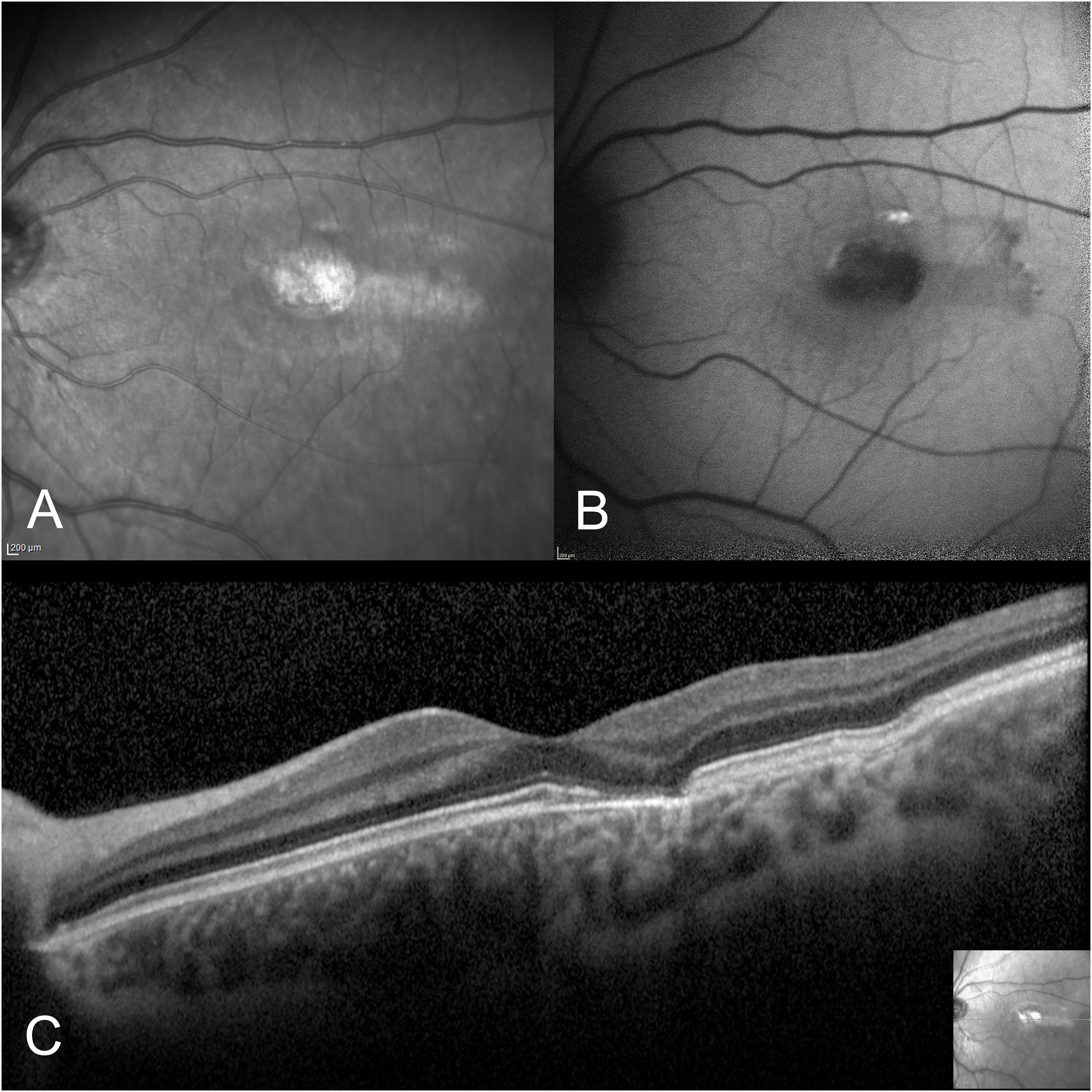
The torpedo-shaped lesion exhibits hyperreflectivity on the infrared reflectance image (A) and appears hypoautofluorescent on fundus autofluorescence (B). Spectral-domain optical coherence tomography (OCT) scan conducted in the region of interest reveals atrophy of the retinal pigment epithelium (RPE) and outer retina, accompanied by heightened posterior hypertransmission. Fundus autofluorescence and OCT images were obtained using the Spectralis HRA + OCT device (Heidelberg Engineering, Heidelberg, Germany).
The optical-coherence tomography (OCT) scan over the lesion evidenced disruption of the myoid, ellipsoid, and interdigitation zones with a barely visible outer limiting membrane, thinning of the retinal pigmented epithelium (RPE) band, hypertransmission of the choroid and subsidence of the overlying retinal layers. The temporal tail of the lesion showed intact ellipsoid zone and localized inner choroidal excavation without subretinal fluid (Figure 2C). The subfoveal choroidal thickness measured 376 μm.
The fluorescein angiography revealed a torpedo-shaped hyperfluorescent area (window defect due to RPE and outer retina damage) ringed with a hypofluorescent rim and accompanied by a faintly hyperfluorescent tail. The central hyperfluorescence appeared in the early angiogram phases and persisted nearly unchanged until the late phases, for the staining of the lesion (Figure 3A).
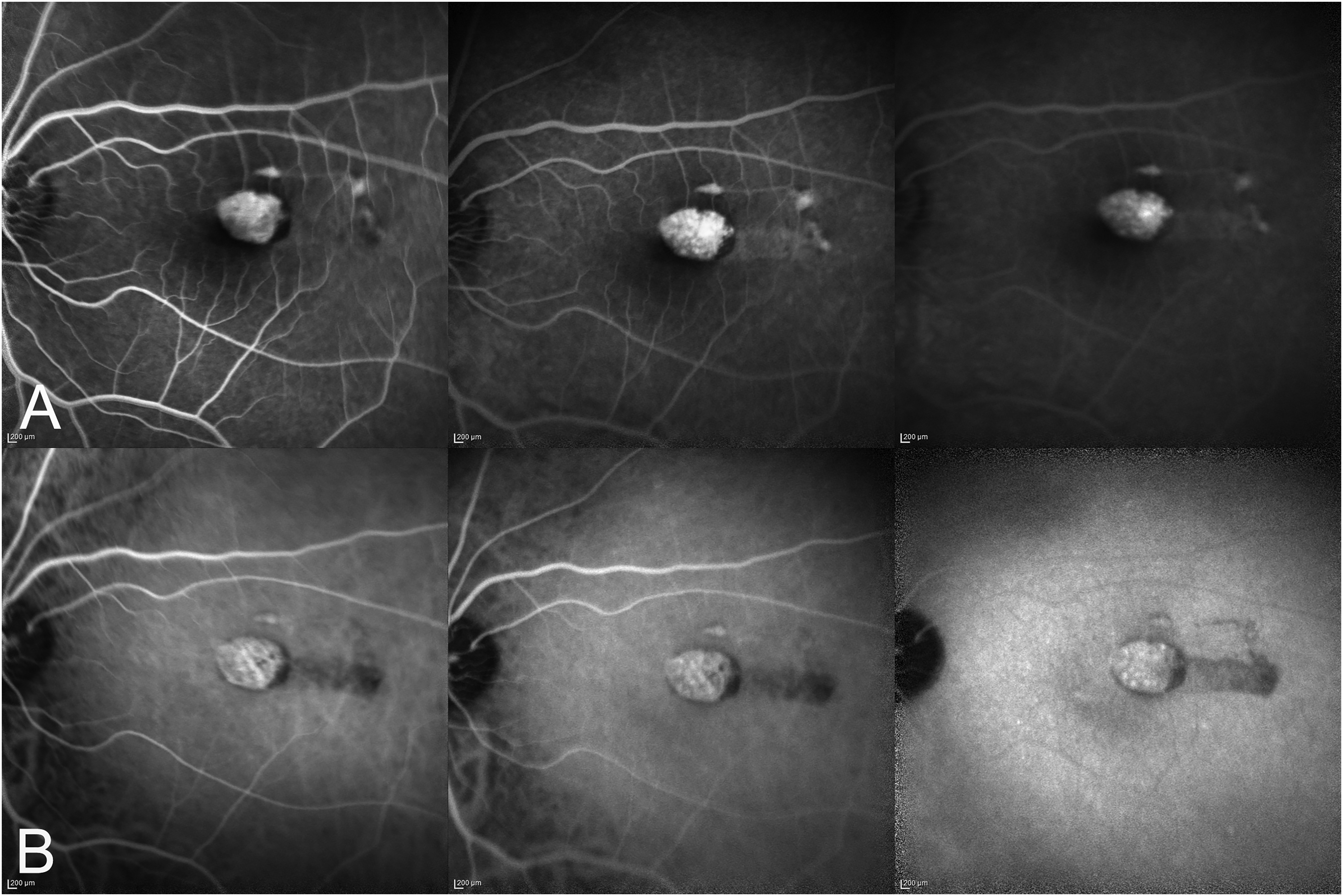
The early, intermediate, and late phases of fluorescein angiography (at times 00:32, 02:06, and 04:38, respectively) (A) and indocyanine green angiography (at times 02:24, 05:43, and 16:07, respectively) (B) do not reveal any significant increase in the size or intensity of fluorescence over time. Both fluorescein angiography and indocyanine green angiography were performed using a Spectralis HRA + OCT device (Heidelberg Engineering, Heidelberg, Germany).
The indocyanine green angiography evidenced a relevant hyperfluorescence due to the increased transmission of the choroid signal during all phases, with the large choroidal vessels clearly visible in the early and intermediate phases. A hypofluorescent wake, looking like a comet tail, was evident until the late angiogram phases (Figure 3B).
The OCT angiography did not show any abnormalities within the superficial or deep capillary plexus. The slab referred to as the outer retina to choriocapillaris (ORCC) slab, extending from the outer boundary of the outer plexiform layer to 8 m beneath Bruch's membrane, revealed the presence of a peculiar vascular pattern compared to the unaffected adjacent choriocapillaris (Figure 4). Convoluted fine vessels were interspersed with flow voids within the head of the lesion, whereas vertically-oriented straight vessels crossed the tail region in a comb-like fashion, alternating with areas of absent flow signal. These vessels represented the anteriorly displaced Sattler's vessels within the lesion's area, due to the outer retina and RPE atrophy along with the choriocapillaris depletion.
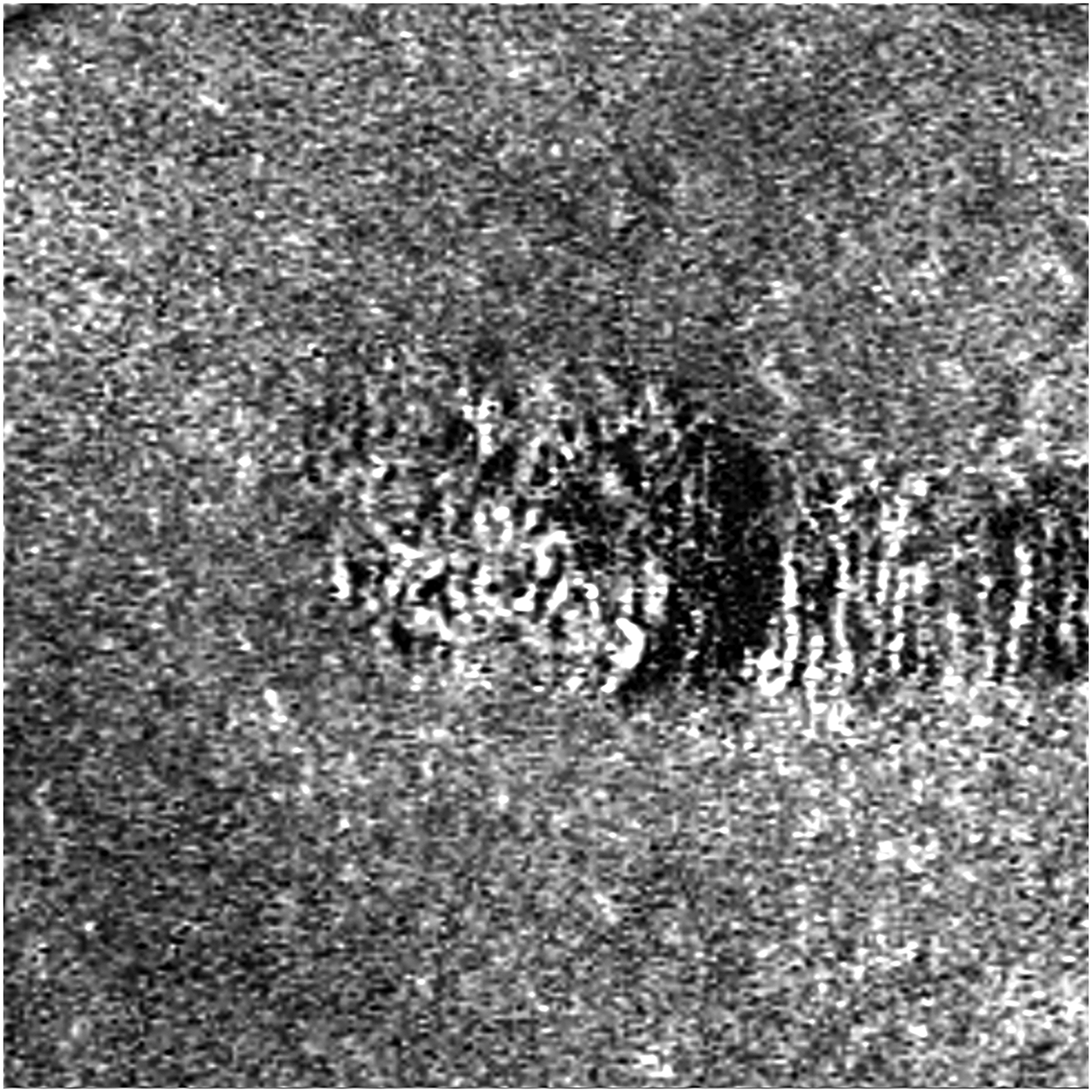
The OCT angiography slab spanning from the outer retina to the choriocapillaris reveals intricate, convoluted vessels with intermittent areas of flow void within the head of the lesion, while vertically-oriented straight vessels are observed in the tail region. The OCT angiography images were acquired and analyzed with ZEISS AngioPlex OCT-Angiography (ZEISS CIRRUS HD-OCT 5000, Carl Zeiss Meditec, Inc., Dublin, USA).
The multimodal imaging ruled out differential diagnoses like chorioretinitis, vitelliform macular dystrophy, other congenital lesions (e.g., CHRPE, chorioretinal scar, coloboma of the macula), and tumors. The diagnosis of type 1 torpedo maculopathy was hence established.
Discussion
We report the first case of TM observed in a patient affected by KC. Although it is difficult to establish a pathogenetic association between the two diseases, it cannot be ignored that KC has been described in association with several posterior segment changes.
In particular, KC has been documented in individuals with Leber congenital amaurosis, 12 retinitis pigmentosa, 13 and retinopathy of prematurity, 14 all of which are diseases associated with the Franceschetti-Leber phenomenon. This distinctive sign, also referred to as the oculo-digital phenomenon, involves the vigorous rubbing of the eyes by children with blindness or low vision, as an attempt to induce visual perception by applying pressure on the eyeball, presumably generating phosphenes. In this context, the connection with KC lies in the mechanical factor of eye rubbing, which predisposes children with visual impairments to the development of corneal ectasia. 15
Additionally, KC may be associated with acquired retinal, optic disc, and choroidal changes.
There are case reports in which KC coexists with conditions such as cuticular drusen, 16 myelinated retinal nerve fiber layer, 17 optic disc pit, 18 central serous chorioretinopathy,19,20 and choroidal neovascularization. 21
Several studies22–27 have more rigorously explored the association between KC and changes in the posterior segment vascularization. It has been found that patients with KC have a thicker choroid compared to healthy subjects.22–27 However, choroidal thickness does not appear to be an independent predictor of KC progression. 28 Histopathological changes in the corneal matrix proteins and collagen, particularly collagen type I, occur in keratoconic eyes, leading to disorganized distribution of collagen fibers. 29 Collagen type I is a key component of vessel walls and changes in collagen fiber patterns might affect blood vessel size. The thickening of the choroid in keratoconic eyes may be explained by its relationship with connective tissue changes. 22 Also inflammatory changes in KC may contribute to increased choroidal thickness, supported by evidence of overexpression of inflammatory mediators in tears and impression cytology samples of affected eyes.22,27
A recent case-control study 27 demonstrated a significant reduction in vessel density within the macular superficial capillary plexus, macular deep capillary plexus, and peripapillary capillaries in the KC cohort when compared to the control group with normal ocular characteristics. Conversely, choriocapillaris flow area was significantly higher in patients with KC than in the control group. According to the authors, the inflammatory processes ongoing in KC eyes induce alterations in the vascular supply, cellular constituents, and chemical milieu of the affected region. These changes, whether transient or enduring, can manifest as discernible structural and functional adjustments also in the tissues of the posterior segment, which can be identified using OCTA. In our young patient, the subfoveal choroidal thickness measured 376 μm, whereas the OCTA showed a choriocapillaris rarefaction paralleled with the anterior displacement of the Sattler's vessels at the level of the lesion, without abnormalities within the superficial or deep capillary plexus. These findings do not appear to align with the vascular changes described in patients with KC in the aforementioned study. To the best of our knowledge, only six reports have described the choriocapillaris changes in TM.30–35 Most of these reports demonstrated a diffuse attenuation of the choriocapillaris signal, supporting the thesis of primary involvement of the RPE/choriocapillaris complex in the pathogenesis of torpedo maculopathy. Papastefanou et al. 32 also assumed a rearrangement of choroidal vessels with the expansion of the Sattler`s layer. Giannakaki et al. 33 mentioned that the Sattler's layer appeared to extend to the lesion-related cleft. On the contrary, Ding et al. 34 found an increased density of the choroidal capillary vasculature, due to enhanced signal transmission through a thinner RPE. Chawla et al. 35 described a convoluted pattern of fine vessels and interspersed empty spaces within choriocapillaris, questioning the assumption that those vessels were from the Sattler's layer.
A noteworthy structural component of the retina to cite is the Bruch's membrane, lying between the RPE and the choriocapillaris, and serving two major functions as the substratum of the RPE and the choroidal vessel wall. 36 This multi-layered membrane has numerous structural components in common with the corneal layers implicated in the pathophysiology of KC, that is corneal stroma, Bowman's layer, and epithelial basal layer.36,37 The structural similarity observed between Bruch's membrane and various corneal components may suggest a potential role for genetic changes in predisposing individuals to these two coexisting abnormalities. Interestingly, in a large genome-wide association study for KC, 36 significant genomic loci implicated in the dysregulation of corneal collagen matrix integrity and cell differentiation pathways have been identified. 38 We cautiously speculate that the combination of diverse genetic variants could have contributed to the patient's susceptibility to developing both keratoconus (KC) and torpedo maculopathy (TM). However, it is important to emphasize that this remains a speculative hypothesis at this stage, providing a preliminary insight into the potential pathogenesis of a rare retinal disorder, such as TM.
In conclusion, this is the first case of TM in a patient affected by keratoconus. Currently, there are insufficient data to explain if the relationship between torpedo maculopathy and keratoconus is causal or fortuitous, and further studies are required to investigate the pathogenesis of this macular disorder.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Summary statement
We present a case of torpedo maculopathy in a young male patient with keratoconus. The diagnosis was made using multimodal imaging. It is uncertain whether this association is based on a common pathogenetic substrate or is purely coincidental. Further studies are necessary to gain new insights into this topic.