
Abstract
Introduction
To report a family with severe ocular disorder caused by double gene variants in causative genes of autosomal dominant cataracts, GJA8 and CRYGC.
Case presentation
A 5-month-old boy with poor vision and enophthalmos was referred to our hospital. Further ocular examination showed horizontal nystagmus, iris abnormalities with pinpoint pupils, and extreme microphthalmia with axial right and left eye lengths of 13.48 mm and 13.75 mm, respectively. Digenic heterozygous variants (c.269T > G, p.Leu90Arg in CRYGC and c.151G > A
Conclusions
To our knowledge, this is the first report of a patient with variants in two cataract-related genes. Importantly, patient with double heterozygous variants in two dominantly inherited genes may suffer more serious phenotypes than those with heterozygous variant in a single dominantly inherited gene. Whole exome or genome sequencing is necessary for a genetic diagnosis in case of multiple gene variants.
Introduction
Congenital cataract, a major cause of vision loss in children worldwide, is responsible for approximately one-third of blindness in infants. The incidence of congenital cataracts varies from 12 to 136 per 100,000 births.1,2 Genetic factors are responsible for approximately 22.3 percent of congenital cataracts. 3 Congenital cataract could be inherited either in isolation or as a part of ocular syndrome or systemic abnormality. It could be accompanied with multi-systemic genetic problems (developmental defects, chronic kidney diseases or metabolic disorders et al.) or anterior chamber developmental anomalies (microcornea, microphthalmia, or aniridia et al.). 4 About 85% of the hereditary congenital cataracts are frequently inherited as an autosomal dominant trait, but could also be inherited as autosomal recessive, or X-linked trait.1,5
In recent years, Next Generation Sequencing (NGS) has been rapidly evolved and adopted in the detection of inherited cataract . The NGS have gradually expanded our knowledge about the spectrum of congenital cataract variants. Up to now, more than 100 genes and 200 loci have been reported to be responsible for congenital cataracts, which could be divided into syndromic and non-syndromic cataract genes.6,7 GJA8 and CRYGC are important and common genes responsible for dominant hereditary cataracts.8,9 In this study, we encountered a boy with double gene variants in both GJA8 and CRYGC and performed a review analysis for GJA8 and CRYGC.
Case description
Informed consent was obtained from the parents of the proband according to the protocol approved by West China Hospital Sichuan University. The procedures in this study conformed to the tenets of the Declaration of Helsinki. We had recruited a family with microphthalmia and anterior segment dysgenesis in this case report. Whole exome sequencing had been performed on the proband's genomic DNA sample. These variants were further confirmed by sanger sequencing and segregation analysis.
A 5-month-old boy with bilateral low vision and enophthalmos was referred to our hospital. The parents in this case report were not from consanguineous marriage and the proband was born naturally at full term (39Weeks). Test of TORCH serologies had been performed to the mother during pregnancy, only immunoglobulin M(IgM) seropositivity of the herpes simplex virus-1 was detected. IgM antibodies of the herpes simplex virus-1 could not cross the placenta and it will not cause cataract to the proband. Additionally, no seropositivity result of the TORCH had been detected in the proband recently. We could rule out the causes of TORCH infections for the proband. However, the boy's parents also suffered congenital serious ocular disorders.
Variants in both CRYGC (c.269T > G (p.Leu90Arg)) and GJA8 (c.151G > A (p.Asp51Asn)) were detected in the proband. These two variants had been predicted to be damaging by prediction of four silico tools (Mutation Taster, GERP++ , MetaSVM, and Provean). The reported variant in GJA8 (c.151G > A, p.Asp51Asn) had been predicted to be likely pathogenic and novel variant in CRYGC (c.269T > G, p.Leu90Arg) was uncertain significant defined by ACMG criteria (https://wintervar.wglab.org/). Sanger sequencing was performed for validation and segregation assessment. Variant in CRYGC (c.269T > G, p.Leu90Arg) was inherited from his affected mother, and the GJA8 (c.151G > A, p.Asp51Asn) variant was inherited from his affected father (Figure 1 and Table 1).
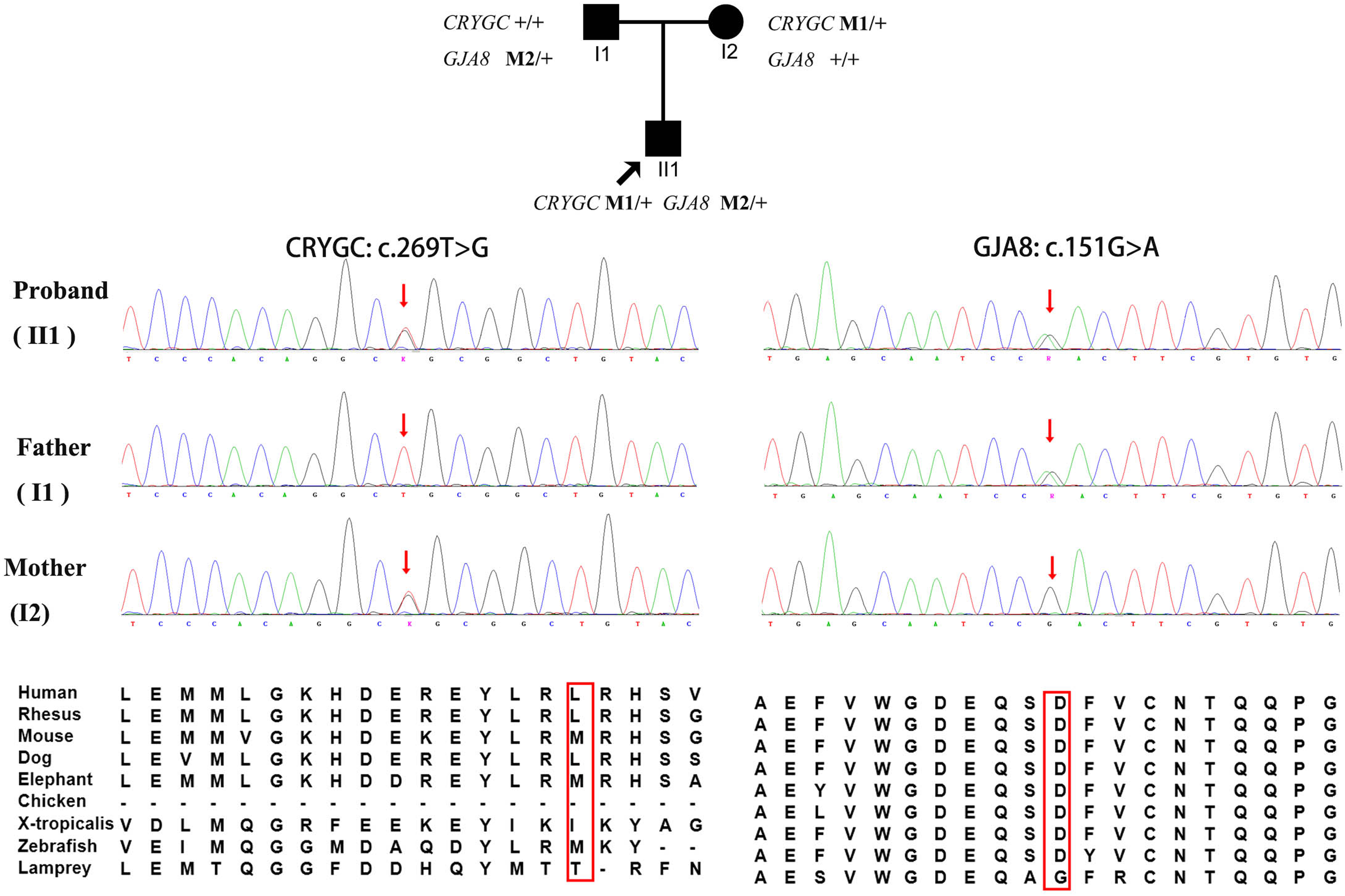
Co-segregation analysis of the proband with variants in both GJA8 and CRYGC. Sanger sequencing and segregation analysis of a family with variants in GJA8 and CRYGC. The first figure shows a family pedigree. Squares and circles indicate males and females, respectively. Black shading indicates affected patients. The arrow indicates the proband. The figures below show Sanger sequencing results revealing variants in GJA8 and CRYGC. Variants in CRYGC (c.269T > G) and GJA8 (c.151G > A) were inherited from the patient's mother and father, respectively. Abbreviation: M1: CRYGC (c.269T > G); M2: GJA8 (c.151G > A).
Clinical data of the family of the patient with variants in GJA8 and CRYGC.

Note: OD, right eye, OS, left eye, BCVA, best corrected visual acuity, AL, axial length, HCD, horizontal corneal diameter, NA, not available, +, yes, -, no.
Extreme microphthalmia, anterior segment dysgenesis (ASD) with pinpoint pupil, and enophthalmos were observed in the proband. Axial lengths (ALs) of the proband's right and left eyes were 13.48 mm (OD) and 13.75 mm (OS). The horizontal corneal diameter (HCD) was 5.5 mm in both eyes. Crypts of Fuchs and folds were missing bilaterally in front of the iris. Furthermore, a needle-like pupil and short anterior chamber depth were observed in the proband. The patient's guardian did not consent to other examinations involving sedative and hypnotic pharmaceutical drugs due to the patient's young age (Figure 2 and Table 1).
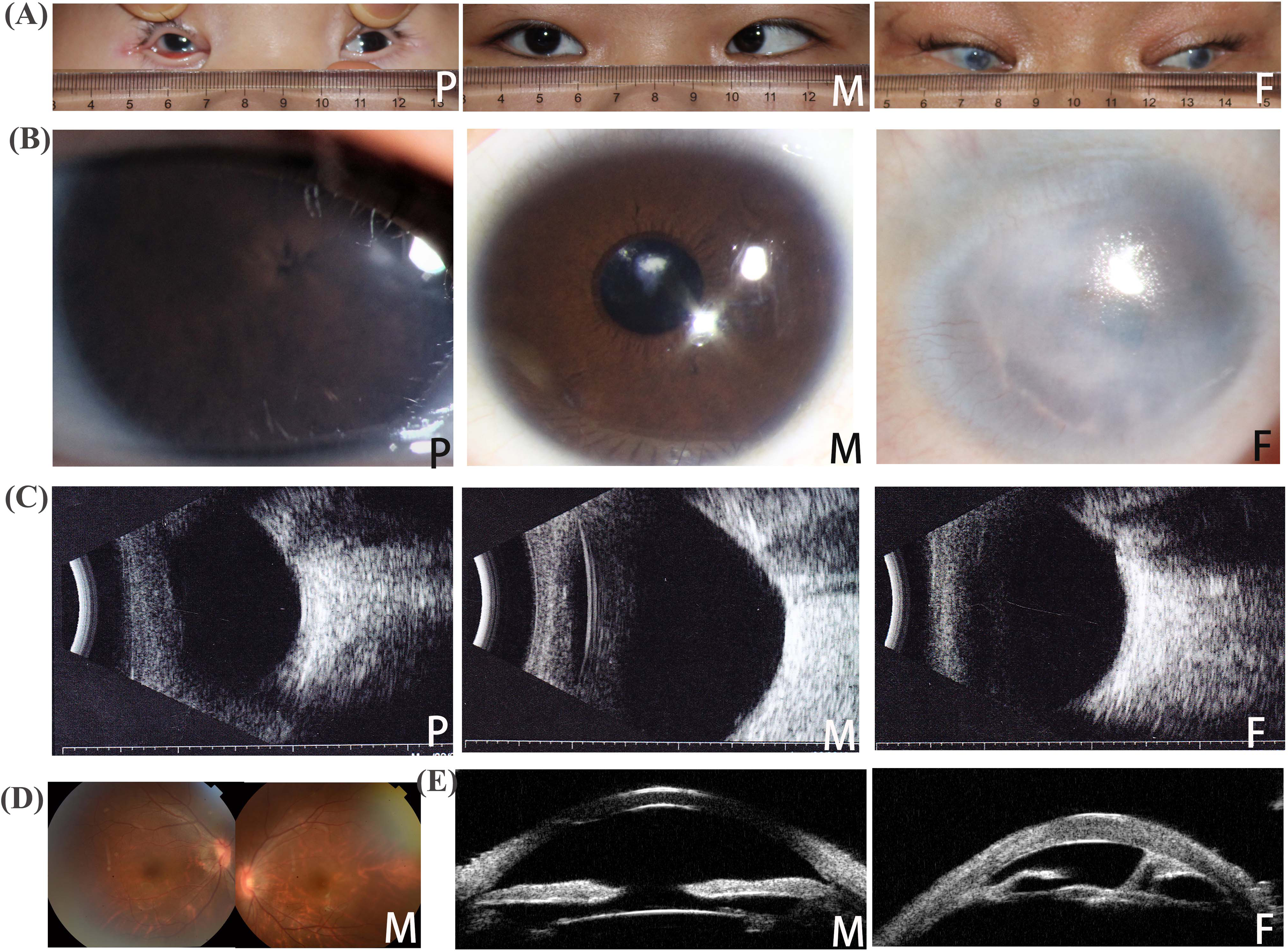
Phenotype of the family of the proband with a double gene variant in GJA8 and CRYGC. A. Photographs of the eyes of each family member. Extreme microphthalmia is observed. B) Photograph of the anterior segment of each family member. Anterior segment dysgenesis is observed. C: B-scan ultrasonography findings of each family member. D: Photograph of the fundus of the patient's mother. E: Ultrasound biomicroscopy findings of the patient's mother and father. Abbreviations: P, proband; M, mother; F, father.
The patient's mother was a 19-years old woman with congenital cataract, microcornea, nystagmus, and strabismus. She was able to count fingers from a distance of 30 cm with the left eye, and her best-corrected visual acuity was 0.03 (Decimal scale) in the right eye. Phacoemulsification combined with intraocular lens implantation was performed in both eyes when she was aged 16 years. Her HCD was 9 mm in both eyes, AL was 23.06 (OD) and 24.90 (OS), and intraocular pressure was normal. No obvious abnormity was detected in the fundus of the patient's mother. The patient's father was a 29-year-old man with congenital cataract, microphthalmia, sclerocornea, glaucoma, and nystagmus. He was capable to count fingers at a distance of 10 cm with the right eye and 20 cm with the left eye. His HCD was 7 mm, bilaterally, and AL was 18.73 mm (OD) and 20.54 mm (OS). The lens thickness of the patient's father was 1.68 mm (OD) and 1.91 mm (OS). Congenital corneal opacification and nystagmus was detected in both eyes. Anterior and posterior adhesion of the iris, angle closure, atrophic ciliary body, rudimentary len, lamellar cataract, and glaucomatous cupping of the optic disk were detected based on ultrasound biomicroscopy, and B-scan ultrasonography (Figure 2 and Table 1).
Discussion
In our study, a 5-month-old boy with poor vision and enophthalmos was detected double heterozygous variants in two dominantly inherited genes (c.269T > G p.Leu90Arg in CRYGC and c.151G > A p.Asp51Asn in GJA8). Horizontal nystagmus, iris abnormalities with pinpoint pupils, and extreme microphthalmia could be detected by further ocular examination. The phenotype of the proband with digenic variants in CRYGC and GJA8 have a more serious phenotype than his parents with a single variant in CRYGC and GJA8, respectively.
Additionally, we performed a genotype-phenotype review of GJA8 and CRYGC based on Cat-Map (https://cat-map.wustl.edu/). GJA8 is a lens gap junction protein and encodes connexin 50 (CX50). CX50 is a component of gap junction channels and functions in a calcium and pH-dependent manner. It had reported that CX50 was involved in the lens growth and maturation of lens fiber cells. Variants in GJA8 are responsible for microphthalmia, microphakia and congenital cataracts. 10 Additionally, CX50 are also detected in the endothelium, fibroblasts, and epithelium of the cornea. It has been known to cause corneal opacifications, sclerocornea. 11 A total of 74 variants in GJA8 were detected in 108 probands with hereditary eye diseases. 74 variants in GJA8 consisted of 70 missense variants, two frameshift deletions, one nonsense variant, and one in-frame deletion. Additionally, six of the 74 variants in GJA8 were homozygous. Among patients with heterozygous variants, 97 (95.10%) were diagnosed with cataract, 15 (14.71%) with microcornea, 11 (10.78%) with microphthalmia, eight (7.84%)with glaucoma, seven (6.86%) with nystagmus, six (5.88%) with coloboma, three (2.94%) with pupil defects, three (2.94%) with myopia, three (2.94%) with corneal opacification, two (1.96%) with anterior segment dysgenesis, two (1.96%) with sclerocornea, two (1.96%) with aphakia, one (0.98%) with microlentis, one (0.98%) with microcephaly, one (0.98%) with strabismus, one (0.98%) with buphthalmos, and one (0.98%) with photophobia. Additionally, the variant in GJA8 detected in our proband (c.151G > A, p. Asp51Asn) has been reported in four affected patients from three families. Bilateral cataracts and microphthalmia were detected in all affected family members.12,13 Sclerocornea, buphthalmos, corectopia, nystagmus, and anterior segment dysgenesis were also found in patients with the same variant (c.151G > A, p. Asp51Asn) in GJA8 (Figure S1).
Mammalian lens crystallins are necessary for maintaining the transparency of lens and could be divided into α-, β-, and γ-crystallins families. The γC-crystallin (the encoded protein of CRYGC) is the most common type of γ-crystallins. CRYGC encodes crystallins which is expressed in the early stage of development, and is abundant in the nucleus of the lens. A total of 33 variants in CRYGC were detected from 44 probands with hereditary eye diseases. These 33 rare variants consisted of 13 missense variants, 11 frameshift deletions, 9 nonsense variants. For the patients with heterozygous variant in CRYGC, 100.0% patients were diagnosed with cataract, 10 (22.22%) with microcornea, 10 (11.11%) with microphthalmia, 3 (6.67%) with glaucoma, 3 (6.67%) with nystagmus, 3 (6.67%) with iridocoloboma, 1 (2.22%) with myopia, 1 (2.22%) with corneal opacification, 1 (2.22%) with aphakia, 1 (2.22%) with optic disc coloboma (Figure S1). Comparing the phenotype of the GJA8 and CRYGC, no significant difference could be detected (p = 0.70). Cataract and microcornea were the most common manifestations of the patients with variants in GJA8 or CRYGC. While significant difference of the variant type could be detected in GJA8 and CRYGC (p = 3.81E-10). Missense was the most frequent variants in GJA8. However, missense and frameshift deletion were the most common pathogenic variants in CRYGC. The variant in CRYGC (c.269T > G, p.Leu90Arg) detected in the proband and mother was not reported before. The proband's parents, who had single gene variants, had phenotypes consistent with those reported in previous studies. The proband, who had a double gene variant in both CRYGC and GJA8, suffered more serious disease manifestations that included extreme microphthalmia. We cannot rule out the impact of each gene defect on the phenotype. The co-occurrence of pathogenic variants in multiple genes is not rare and mainly depends on the frequency and mode of disease inheritance.14,15 For example, in patients with breast cancer, the coexistence of variants in BRCA1 and ATM, BRCA1 and BRCA2 variants have been previously reported, although not widely.16,17 Patients with double heterozygous variants in different genes develop breast cancer at an earlier age, have more severe disease, and are more likely to develop multiple primary tumors than single heterozygous carriers.16,18,19 Additionally, heterozygous variants in ATM in BRCA1 knock-out mice increase the severity of mammary gland cancer and reduce ductal branching, suggesting a synergistic interaction between both proteins with respect to tumorigenesis. Subsequent in vitro studies indicate that double heterozygosity in ATM and BRCA1 leads to increased cell transformation rates versus single heterozygosity. 17 Variants in two distinct genes have also been reported in inherited ocular disorders. Double de novo heterozygous ATP2A2 and PAX6 variants have been reported in patients with a unique combination of Darier disease, multiple bone cysts, and bilateral aniridia. 14 Double heterozygosity in FRMD7 and GJA8 genes has been linked to the pathogenesis of congenital nystagmus and cataracts. Patients with double variants in different genes could be responsible for more severe phenotypes. 20
If screening for a single gene had been performed to diagnose the patient, the pathogenic variant would likely have been missed. This could result in a serious medical malpractice investigation if used to obtain a prenatal diagnosis.
Conclusion
In conclusion, patients with double heterozygous variants in two dominantly inherited genes may suffer more serious phenotypes than those with heterozygous variant in a single dominantly inherited gene. While the interaction between the genes causing the phenotype seen, the mechanism is unclear and needs further study. Whole exome or genome sequencing is necessary for the genetic diagnosis of patients with multiple gene variants.
Supplemental Material
sj-docx-1-ejo-10.1177_11206721231163611 - Supplemental material for Microphthalmia and anterior segment dysgenesis due to a double gene variant in GJA8 and CRYGC
Supplemental material, sj-docx-1-ejo-10.1177_11206721231163611 for Microphthalmia and anterior segment dysgenesis due to a double gene variant in GJA8 and CRYGC by Lin Zhou, Ganghua Wang, Bin Hu, Hui Jiang, Fanwen Jiang and Zhuping Xu in European Journal of Ophthalmology
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the 135 project for disciplines of excellence-Clinical Research Incubation Project, West China Hospital, Sichuan University, Natural Science Foundation of Sichuan Province, Chengdu Science and Technology Program, (grant number No.2021HXFH026, 2022NSFSC1370 , 2021-YF09-00024-SN)
Informed consent statement
This case report was based on the clinical data of the patient and had been approved by West China hospital, Sichuan university. Written informed consent followed the tenets of the Declaration of Helsinki had been obtained from the proband and family members.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.