
Abstract
In this report we illustrate the ophthalmologic assessment of two patients affected by Proteus Syndrome (PS), an extremely rare genetic disorder. Case #1 describes a 26 year old male patient followed for multiple ophthalmic anomalies: a limbal dermoid cyst, a unilateral cataract, bilateral nystagmus, severe myopia and unilateral optic nerve head drusen. Case #2 describes a 20 year old female patient referred to our Ophthalmology Department for a routine ophthalmologic evaluation after being treated for 3 years with Miransertib (an experimental AKT-pathway inhibitor). Both patients underwent a complete ophthalmologic examination and a multimodal imaging evaluation. The multimodal imaging approach has revealed useful to evaluate both cases in detail and to keep track of disease evolution over time, moreover providing helpful features to further characterize this rare syndrome.
Introduction
Proteus Syndrome (PS) is an extremely rare disease, with approximately 100 cases known in literature. 1 This polymorphic, hamartomatous/neoplastic disorder is caused by a post-zygotic somatic activating mutation in the oncogene AKT1 (a serine-threonine protein kinase) 2 and it is characterized by abnormal segmental overgrowth involving different tissues and organs (eg: connective tissue, bone, muscular tissue, vascular tissues). 3 Onset usually occurs at birth or early in childhood (6–18 months). Definitive diagnostic criteria require specific clinical features supported by a genetic confirmation of AKT1 mosaicism. 1 The missense mutation in the AKT1 gene [c.49G>A; p.(Glu17Lys)] detected in affected tissues is distinctive for PS, allowing the differential diagnosis with other similar overgrowth syndromes.1,4
In this report we describe two patients with a genetically confirmed diagnosis of PS, evaluated with a comprehensive ophthalmological examination, orthoptic assessment and multimodal imaging studies.
Patients provided written informed consent for publication of their clinical cases and related images.
Case #1
26 years old male patient born from non-consanguineous parents after an uncomplicated, full-term pregnancy and delivery. Family history was unremarkable. Clinical diagnosis of PS 3 was prompted at 2 year of age and confirmed by AKT1 gene mutation at 20 years old.
Patient history was remarkable for mild intellectual and motor disability, deep venous thrombosis and orthopedic issues (such as limbs asymmetric overgrowth, scoliosis, cranial hyperostosis); he underwent knee and heel surgery, nasal polypectomy, uvula papilloma resection and parotid pleomorphic adenoma resection.
The patient was referred to our Ophthalmology Department in 2012 (aged 15 years old) with an history of high myopia and amblyopia in the right eye (RE), treated with occlusion therapy and use of corrective lenses since he was 6 years old. Since 2012 he has been examined annually with a complete ophthalmological and orthoptic assessment.
At last examination in 2021 the best corrected visual acuity ( [Right Eye] 1.7 logMar with −10sph −2.75 cyl(30°) [TABO] [Left Eye] 0.15 log Mar with −3sph −3 cyl(170°) [TABO]
BCVA didn't show major changes in both eyes compared to first visit in 2012.
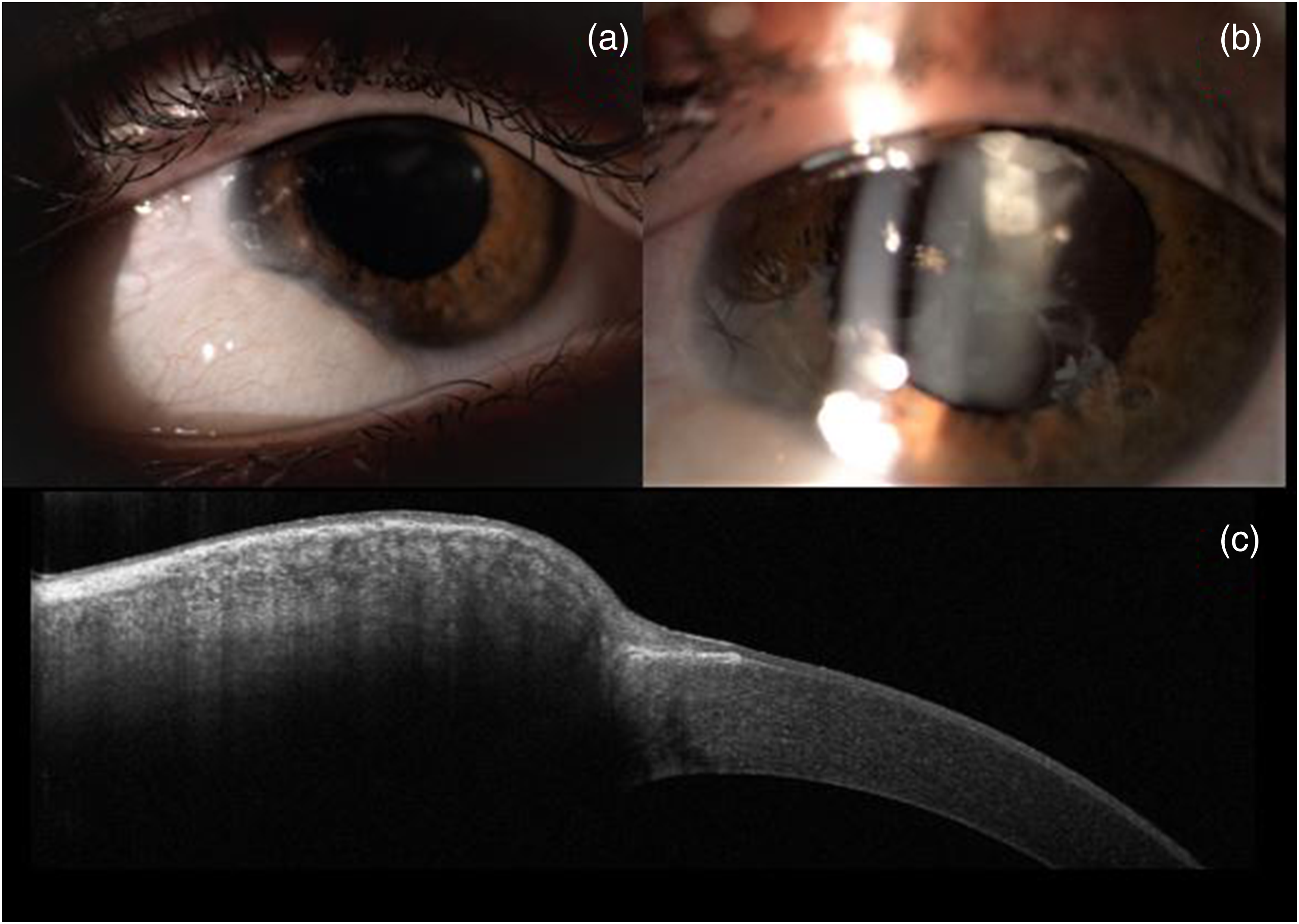
(a) Limbal dermoid cyst in the inferotemporal quadrant in RE; (b) Peripheral superior cortical lens opacity with pigment on anterior surface of the lens in RE; (c) AS-OCT scan on the limbal dermoid cyst.
Acquisitions via
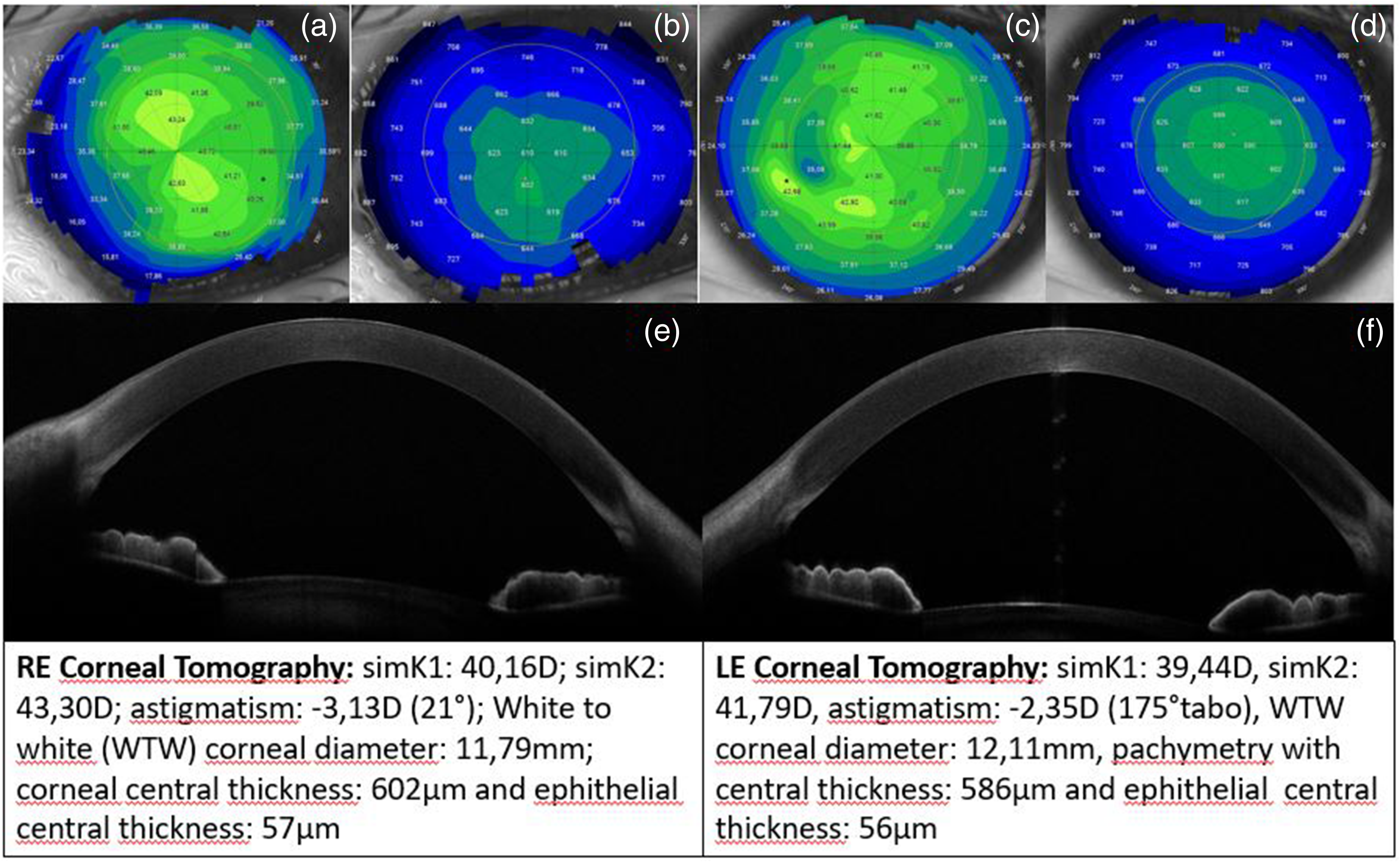
(a-b) RE tangential corneal tomography and pachymetry (with detected parameters); (c-d) LE tangential corneal tomography and pachymetry (with detected parameters); (e) RE anterior segment OCT with normal features, except for temporal margin where a limbal dermoid cyst is detected; (f) LE anterior segment OCT with normal features.
RE fundoscopy showed severe myopic retinocoroidosis, abnormal macular reflex, multiple pigmentary changes and an optic nerve head with slight elevation, blurred margins and greyish colours attributable to optic nerve drusen (Figure 3 – a). LE fundoscopy revealed an unremarkable retina, except for mild tilted optic disc. (Figure 3 - b)
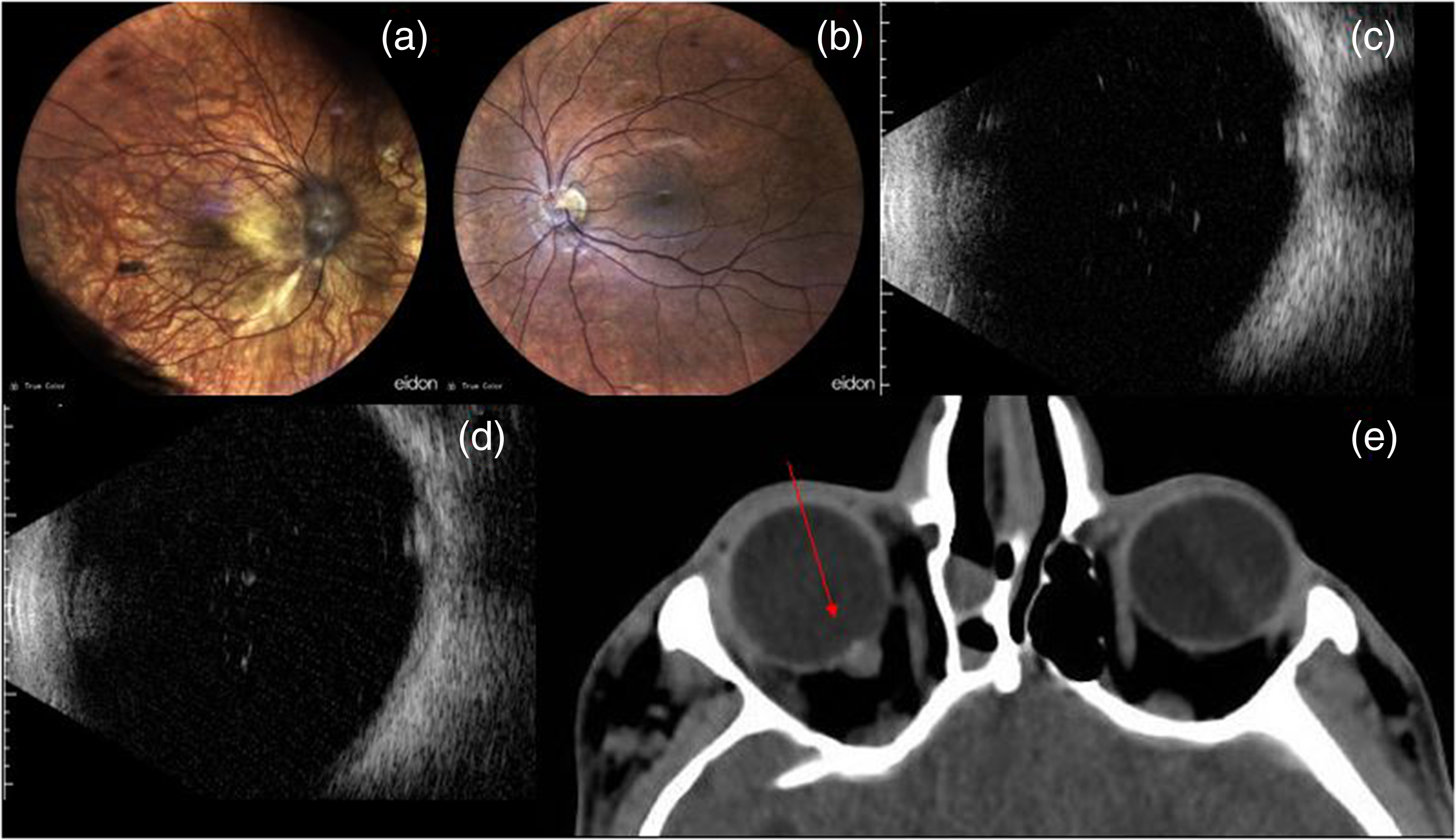
(a) RE fundus: severe myopic retinal-coroidosis and optic nerve drusen; (b) LE fundus: unremarkable retina, except for mild tilted optic nerve; (c-d) RE ultrasound B-Scan in 2013 and 2019: elevation of the optic nerve head (ONH) with medium reflectivity due to slightly calcified drusen; (e) Cranial CT-scan: ONH in RE slightly elevated and hyperintense compatible with mildly calcified drusen.
During last 10 years follow-up, three
Multiple
Macular and optic nerve Optical Coherence Tomography (OCT - Cirrus 6000/Zeiss) acquisition was attempted unsuccessfully due to nystagmus and head mispositioning.
Case #2
20 years old female patient, born from non-consanguineous parents after an uncomplicated, full-term pregnancy and delivery. Family history was unremarkable. Progressive hand overgrowth was noticed soon after birth, prompting clinical diagnosis of PS, 3 subsequently confirmed by AKT1 gene mutation identification.
The patient had several musculoskeletal issues, such as severe rotoscoliosis and hyperostotic fusion of all cervical vertebrae, progressive hands overgrowth (treated with amputations of fingers) and right tibial anomalies. Moreover, she showed portal cavernomatosis complicated by partial thrombosis of the extrahepatic veins and developed, at 13 years old, a low-grade serous carcinoma of the ovary (LGSOC) treated with multiple surgeries and personalized target therapy with Miransertib. 5
The patient was referred to our Ophthalmology Department for a routine ophthalmologic examination to evaluate possible ophthalmic complications related to Miransertib, an experimental pan-AKT inhibitor assumed daily over 3 years by the patient.
Her ophthalmic history was unremarkable. Uncorrected best visual acuity (
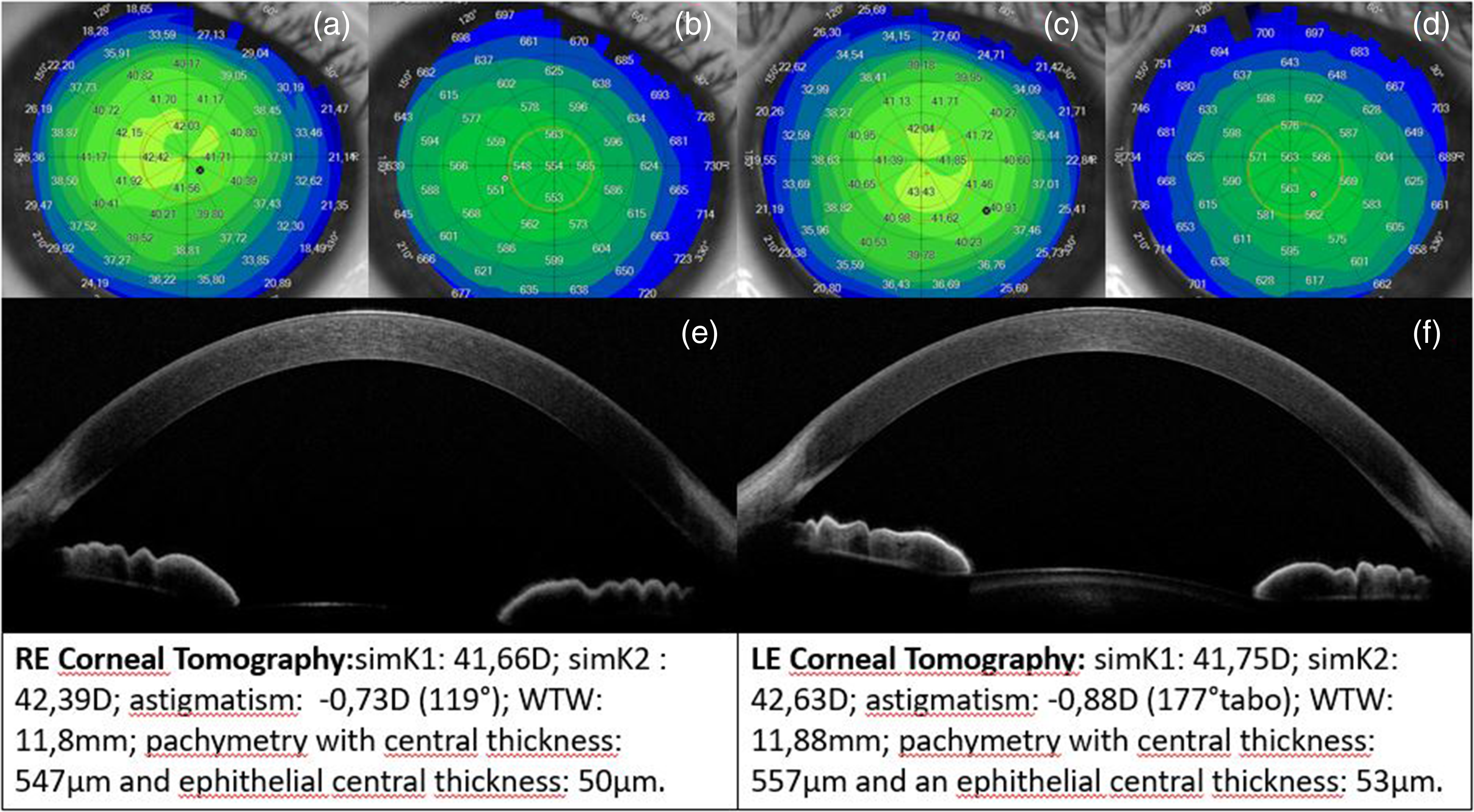
(a-b) RE tangential corneal tomography and pachymetry (with detected parameters); (c-d) LE tangential corneal tomography and pachymetry (with detected parameters); (e-f) RE and LE Anterior Segment OCT with normal features.
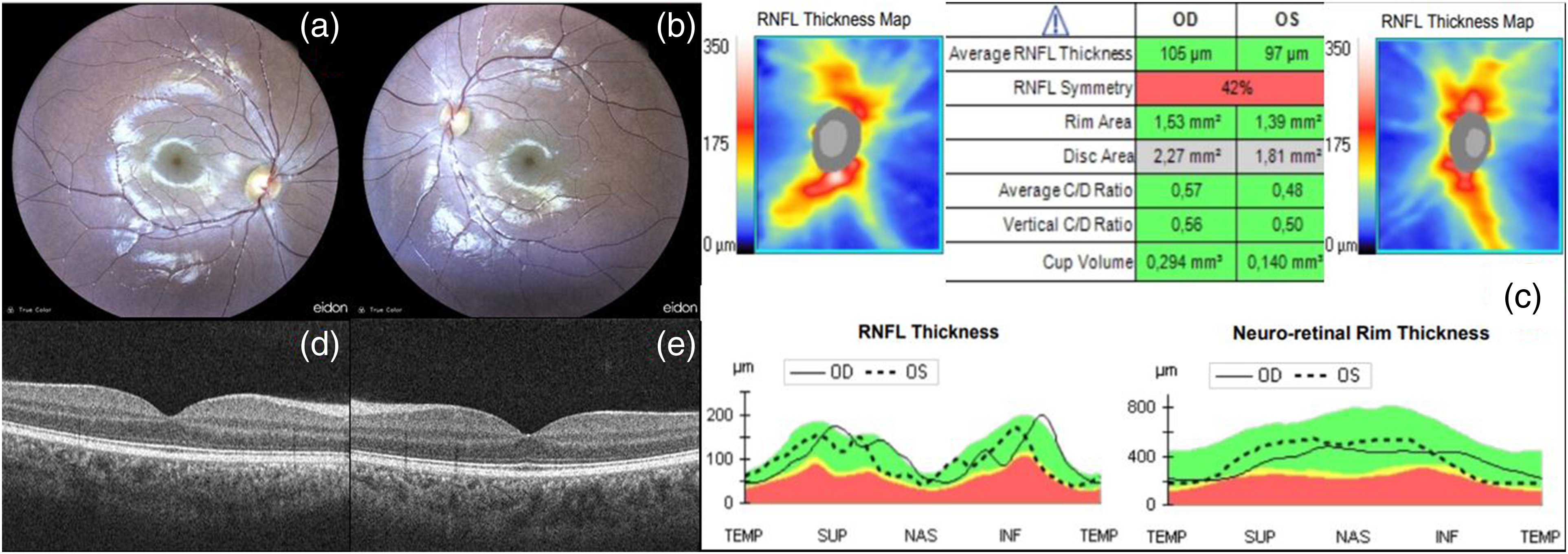
(a-b) RE and LE fundus with unremarkable retina; (c-f-g) ONH and RNFL OCT with normal feature in both eyes (except for RNFL symmetry); (d-e) RE and LE horizontal macular scan: physiological conformation of all macula layers.
Conclusion
In current medical literature various reports have described the ophthalmic manifestations associated with PS 6 ; however, many studies have been carried out before the introduction of the new diagnostic criteria of PS, which require genetic confirmation of AKT1 gene mutation (besides common clinical features). Only about 47.3% of the published reports meet the current diagnostic criteria; therefore, many papers may misdiagnose PS with other overgrowth syndromes. 7
This article describes the ophthalmic evaluation of two genetically confirmed PS patients with a thorough multimodal ocular imaging assessment for the first time.
PS arises from a post-zygotic somatic mosaicism, due to a missense activating mutation in the AKT1 oncogene, causing disproportionate overgrowth of affected tissues. In some patients ocular tissues may also be involved, 1 explaining the high frequency of epibulbar tumours, high myopia and morpho-functional alterations of neuro-retinal layers (due to disruption of retinal PI3K-AKT1 signalling pathway). 8
Case #1 presented different ocular alterations related to PS, affecting mostly the RE: a dermoid cyst, a cortical cataract, high myopia (with related retinal morphologic alterations) and optic nerve head drusen; LE, instead, was substantially spared. The asymmetry in eyes involvement is a feature strongly associated with PS, where a hemi-hypertrophy of the body is often found; we suppose it may be related to a somatic mosaicism of AKT1 gene selectively affecting the development of optic grooves and optic vesicles from the neuroectoderm, in an unilateral fashion, during early eye's embryogenesis. Despite PS is a progressive overgrowth syndrome, case #1 patient didn't show any relevant clinical ophthalmic worsening during our 10 years follow-up, nor alterations in subsequent ocular imaging acquisitions.
In contrast to case #1, case #2 didn't show any ocular manifestation of PS, likely due to an absence of AKT1 mutation during early embryogenesis of both eyes.
Multimodal imaging has revealed useful in both cases. In case #1 SA-OCT and Corneal Tomography allowed with an accurate sizing to rule out dermoid's progression and complications. An enlarging lesion could erode limbal junction or induce an irregular astigmatism, both beeing indications for a surgical treatment. Likewise, ECO and CT scan were diagnostic for ONH drusen; explained ONH morphology and excluded the endocranial hypertension or orbital neoplasm. Finally, colour fundus acquisitions enable to keep track of macroscopic progression and show other physicians how could appear retina of a Proteus patient, especially considering the asymmetric involvement with an healthy controlateral eye.
Case #2 didn't show an obvious clinical involvement; nevertheless, multimodal imaging approach investigate possible microstructural alterations and improves our knowledge on this rare syndrome. Furthermore, case #2 supported a good ocular safety profile to Mirasentib. This is an encouraging point: beeing this drug the only to inhibit the AKT1 oncogene activation, hopefully it could be implemented in future to slow ocular disease progression when needed.
Some imaging acquisitions have been difficult to perform in both patients, because of operator-independent factors. Case #1 presented a frontal exostosis (which hindered correct head positioning) and severe nystagmus (greater in RE than LE), preventing to carry out a macular and ONH OCT scan; of note, this acquisition would have been helpful to evaluate ONH drusen and eventual foveal misalignment, a recently described finding in PS patients. 7 Case #2 presented a non-reducible left head tilt (due to hyperostotic fusion of cervical vertebrae), that explain the rotation of the eyes visible in the acquisitions.
This report also supplies evidence regarding the ophthalmic safety profile of Miransertib.
Miransertib (ARQ 092) is an orally-bioavailable, selective, allosteric pan-AKT inhibitor that targets both the active and inactive forms of AKT1, AKT2, AKT3 and AKT1-E17K mutant, by inhibiting phosphorylation of AKT. 9 In Proteus patients, phosphorylation of Ser473 and Thr308 on AKT causes constitutive activation of AKT/PI3K pathway, mediating processes including increased cell proliferation and decreased apoptosis; this accounts for widespread connective tissues overgrowth as well as the pathogenesis of some forms of cancers. Recent studies suggest an overall improvement in symptoms and quality of life of PS patients treated with Miransertib, by slowing or arresting uncontrolled tissues” growth.10,11
Case #2 patient developed a LGSOC, and genetic analysis demonstrated AKT1-E17K gain-of-function mutation on affected connective tissues and ovarian tissue. Considering the benefits of AKT-inhibitors in both PS patients and AKT-mutant cancers, the patient was treated with Miransertib on a compassionate basis for more than 3 years. During this period, Miransertib was well systemically tolerated and in our ophthalmic evaluation no ocular alterations related to this therapy were detected.
Future studies should focus on enrolling patients with a genetically confirmed diagnosis of PS to update the knowledge of this disorder. As concern the application of multimodal imaging in this syndrome, it has proven useful in both cases to thoroughly examinate ocular involvement and to trace the possible progression of the disease over time; a further use of this approach could deepen the knowledge about the microscopical and histological alterations of ocular tissues and structures in PS patients. We conclude by suggesting a complete ophthalmologic and orthoptic assessment in all patients affected by PS, comprehensive of multimodal imaging acquisitions and followed by regular checkups, to evaluate ocular involvement in detail and to keep track of eventual disease progression over time.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article
Research ethics and patient consent
The study was conducting according to the principles of the Declaration of Helsinki and all patients signed an informed consent form.