
Abstract
Purpose
Compare 0.30% sodium hyaluronate (0.30%HA) ocular gel with 0.18%HA eye drops in terms of improvement of ocular signs and symptoms, in patients with moderate to severe dry eye disease (DED).
Methods
This was a multicentric, randomized, investigator-masked, non-inferiority, comparative study conducted over 84 days. Three visits were scheduled, testing fluorescein corneal and conjunctival staining (Oxford and Van Bijsterveld scores), tear film break-up time (TBUT), Schirmer test, DED symptoms, 5-Item-Dry-Eye-Questionnaire (5-DEQ), patient and investigator satisfaction and frequency of instillation.
Results
At Day 35 (D35) and Day 84 (D84), both groups (n = 35 each) had a significant improvement in corneal staining (p < 0.001) with no inter-group difference. Van Bijsterveld score improved earlier (D35) for 0.30%HA suggesting a faster effect on conjunctival epithelium healing. There was no difference between the two concentrations in terms of TBUT or Schirmer improvements; however, the Schirmer test increase was only significant for 0.30%HA at D35 (p = 0.040). At D35 and D84, both groups showed similar improvements of DED symptoms and DEQ-5 score. Furthermore, treatment satisfaction was similar for the 2 formulations suggesting that daily use of 0.30%HA do not cause gel-related blurred vision disturbances. Frequency of instillation was similar for both groups.
Conclusion
Our study demonstrates the non-inferiority of 0.30%HA gel compared to 0.18%HA solution in patients with moderate to severe DED. Because of its gel formulation and higher HA concentration providing prolonged comfort without causing visual disturbances, 0.30%HA gel might be adapted for bedtime use or during the day in more severe conditions.
Introduction
According to the newest report of the Tear Film and Ocular Surface Society's International Dry Eye Workshop (TFOS DEWS II) published in 2017, Dry Eye Disease (DED) is a multifactorial ocular surface disease characterized by a loss of homeostasis of the tear film that results in symptoms of discomfort, irritation and visual disturbance. Many factors have been identified to play a key role in the pathogenesis of this disease such as tear film instability, tear film hyperosmolarity, inflammation and ocular surface damage. 1 Neurosensory abnormalities have been recently added to this panel and can explain the discrepancy often reported between patients’ symptoms and the clinical signs seen on examination. 2
Even though DED is underdiagnosed, it remains a commonly occurring condition seen routinely in everyday clinical practice. A population-based study conducted in the Unites States estimated the annual incidence of DED to be 0.87% in 2012, making it third in ranking of ocular conditions with the highest incidence, after disorders of refraction and/or accommodation and cataract. DED was also the only ocular condition in constant increase each year from 2008 to 2012. 3 Symptoms of DED are often associated with significant psychosocial burden and reduced quality of life (QoL) and can severely impact important daily activities and productivity at work. 4
Hyaluronic acid (HA) is an anionic glycosaminoglycan with a viscoelastic rheology that has been widely used in lubricant eye drops in recent decades. By effectively binding water and resisting dehydration, HA improves lubrication of the ocular surface, increases tear stability, promotes epithelium healing and ameliorates the intensity of dry eye symptoms. 5 Treatment with HA-only formulation topical drops has also been proven to increase QoL scores and patient satisfaction, especially in mild-to-moderate DED.4,6
Higher HA concentrations (0.30%) have proven to be more effective than 0.10% and 0.18% HA in experimental studies on mice. 7 The objective of this study was to compare 0.30% HA gel with 0.18% HA solution in terms of improvement of ocular signs and symptoms, in patients with moderate to severe DED.
Material and methods
Design and procedure
This was a prospective, multicentric, comparative, investigator-blinded, non-inferiority trial (NCT number NCT03645850) that included patients with moderate to severe DED who were already being treated with non-preserved artificial tears in the 3 months preceding the inclusion. Patients were included from 12 European centres: 3 in France, 1 in the United Kingdom, 2 in Poland and 6 in Spain.
After a 7 to 14-day wash-out period with preservative-free (PF) 0.9% NaCl (3–6 drops per day), patients included were randomized into 2 groups receiving either PF 0.18% HA eyedrop solution or PF 0.30% HA gel throughout the follow-up at a dosage of 1 to 2 drops in each eye between 4 and 6 times per day. Patients were identified by a patient number. Each patient number was associated to a treatment number, according to a centre-specific randomization list provided by a statistician before the beginning of the clinical investigation and randomized in either one of treatment groups (allocation ratio 1:1). Both subjects and care providers were blinded to the intervention.
All patients were examined by an ophthalmologist at inclusion (D0) and then reevaluated at day 35 (D35) and day 84 (D84). Corneal and conjunctival staining were assessed on slit lamp examination after instillation of a drop of fluorescein (Oxford score) or lissamine green (Van Bijsterveld score), respectively. Tear break-up time (TBUT) was measured after instillation of a drop of fluorescein as the elapsed time from a normal blink to the appearance of a dry spot on the surface of the eye. Schirmer 1 testing was done without anesthesia and with open eyes. For ocular dryness severity, eight different ocular dryness symptoms were evaluated by the patient at each time point on a scale from 0 (no symptom) to 10 (very important): discomfort, burning, stinging, eye dryness sensation, itching, sandy feeling/foreign body sensation, photophobia and blurred vision. All symptoms’ scores were added to calculate a global total score from 0 to 80. Also, treatment satisfaction and global product performance were evaluated by the investigators and the patients at D35 and D84 on a 4-points scale from unsatisfactory to very satisfactory.
Sample size
The clinical non-inferiority margin of 0.30% HA versus 0.18% HA was set at 2 points on the Oxford score. Considering a standard deviation equal to 2.5, a total of 68 patients (34 patients in each group) was required to reach a power of 90% to set-up the non-inferiority based on a bilateral confidence interval at 95%. Assuming a drop-out rate of 15%, 80 patients were scheduled to be randomized in the study to keep sufficient power for main analysis of the primary end point.
Inclusion criteria
Baseline severity of DED was assessed by the 5-Item Dry Eye Questionnaire (DEQ-5) and by slit lamp examination. Subjects included were 18 years or older and had moderate to severe DED, defined by a DEQ-5 score ≥ 6 with at least one eye having a global ocular staining (cornea and conjunctiva) between 4 and 9 on the Oxford scale8,9 and one of the following criteria: a Schirmer 1 test between 3 and 9 mm/5 min or a mean TBUT ≤ 30 s (sum of 3 consecutive measurements).10,11
Exclusion criteria were pregnant or nursing women; subjects unable to express their consent; contact lens wearers; subjects with severe ocular dryness, eyelid or blinking malfunction, corneal disorders not related to DED, ocular metaplasia, filamentous keratitis or corneal neovascularization; subjects with severe meibomian gland dysfunction; patients with a history of ocular trauma infection or inflammation not related to DED within the last 3 months prior to inclusion; history of ocular allergy or ocular herpes within the last 12 months; ocular surgeries (including laser surgery) in either eye within the last 6 months; other ocular surface diseases; subjects having used artificial tears in the 6 hours preceding the inclusion visit; patients who have received eye drops in either eye with any ophthalmic medication (except for artificial tear substitutes) within 2 weeks prior to study or expected to receive ocular therapy during the study; and patients using any of the following ocular treatments in the month preceding the inclusion: isotretinoin, cyclosporin, tacrolimus, sirolimus, pimecrolimus or punctal plugs.
The investigation was conducted with the ethical principles initially outlined in the Declaration of Helsinki. In accordance with each national regulation, the initial study clinical investigation plan and documents were reviewed and received a favourable opinion of an Independent Ethics Committee. Informed consent was obtained for all patients verbally and in writing before the realization of any specific procedure. No patient was included and/or randomized before having signed the consent form, written in an understandable language.
Efficacy parameters
The primary study endpoint was the comparison of the two HA concentrations in terms of corneal and conjunctival staining (Oxford score) on worse eye between D0 and D35. The worse eye was defined as follows: if only one eye was eligible (i.e. fulfils all ophthalmological inclusion criteria with no ophthalmological exclusion criteria), the study eye was the eligible one; however, if both eyes were eligible, the worse eye was the one with the highest Oxford score, or if not applicable the one with the highest Van Bijsterveld score, or if not applicable the one with the lower Schirmer test results, or if not applicable the one with the lowest TBUT. When all parameters were comparable, the right eye was randomly assigned as the worse eye.
Secondary criteria were also studied on the worse eye at baseline and then reassessed at each follow-up point (D35 and D84). The per protocol sequence of performed procedures at each visit was as follows: evaluation of the average frequency of instillation (collected via a daily-log completed by the patient); collection of adverse events (classified into mild, moderate or severe); evaluation of ocular dryness severity by the patient; evaluation of the DEQ-5 score; evaluation of TBUT; evaluation of the Oxford score; evaluation of Schirmer test; evaluation of the van Bijsterveld score; and evaluation of treatment performance by both the patient and the investigator.
Statistical analysis
Three populations were defined: the Intention to Treat (ITT) population corresponding to all randomized patients; the Safety population corresponding to all randomized patients having received at least one dose of treatment; and the Per Protocol (PP) population corresponding to patients of the Safety population without major protocol deviation (as decided by the Data Review Committee during a blind review process).
Statistical analysis was performed on the worse eye for all participants using SAS© GUIDE software. The main analysis of the main criterion was conducted on the PP population. Results were expressed as means ± standard deviations (SD), and percentages with confidence intervals of 95%. A two-way analysis of covariance (ANCOVA) model was conducted using the main effects of treatment and baseline score as covariates. Adjusted means (least square mean and standard error of the mean) by treatment were presented, as well as an estimate of the difference between adjusted means. A 95% two-sided confidence interval, based on the ANCOVA model, was computed for the difference of 0.30% HA versus 0.18% HA. All statistical tests were assessed at α = 5% level of significance in a bilateral approach.
Results
Among a total of 109 screened patients with informed consent, 81 were eligible for the study and were randomized on D0: 41 patients in the 0.30% HA group and 40 patients in the 0.18% HA group (ITT population). 11 patients prematurely withdrew from the study or were excluded because of screening failure, withdrawal of the informed consent, loss of follow-up or protocol deviation (decided by a Data Review Committee during a blind review process). In total, 70 patients (35 in each group) completed the study throughout the follow-up (PP population).
The mean age of patients included in the PP population analysis was 56.8 ± 11.6 years. Most of the patients included were women (85.7%). Demographics and baseline characteristics of the PP population by treatment group are summarized in Table 1.
Demographics and baseline characteristics of patients with moderate to severe dry eye disease treated with either 0.30% HA or 0.18% HA.

HA: hyaluronic acid; TBUT: tear break-up time.
At D35, both groups had a significant improvement in corneal and conjunctival staining by fluorescein compared to baseline (Oxford score change: −32.8% with 0.30% HA, p < 0.001 and −23.4% with 0.18% HA, p < 0.001) – Figure 1. There was no significant difference between the 2 groups. The sensitivity analysis on the ITT population (with and without replacement of missing values) confirmed this non-inferiority.
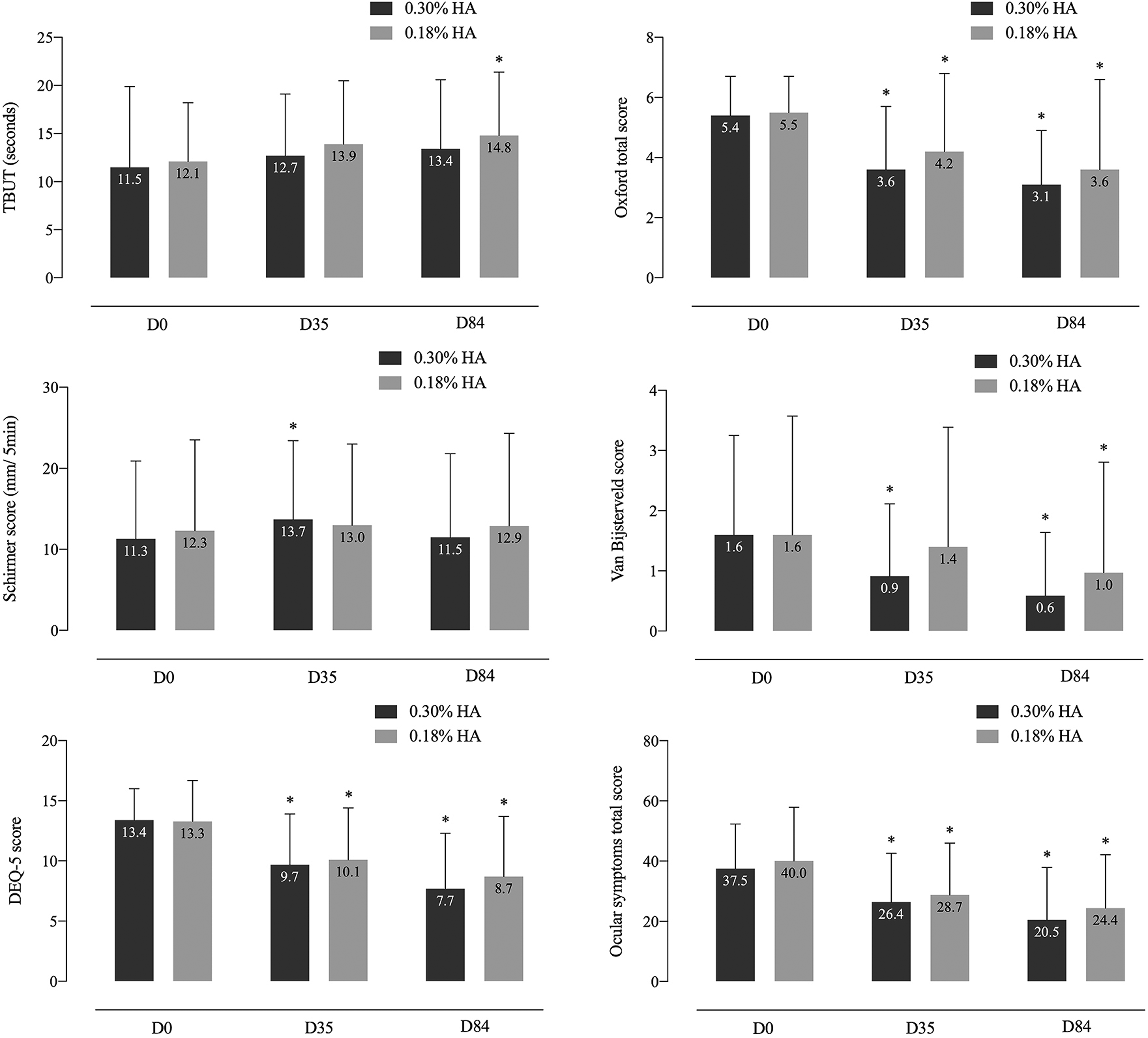
Evolution of tear break-up time (TBUT) and Schirmer, 5-Item Dry Eye Questionnaire (DEQ-5), Oxford, van Bijsterveld and ocular symptoms scores in moderate to severe dry eye patients receiving either 0.30% or 0.18% hyaluronic acid (HA) over 84 days of follow-up.
TBUT was slightly but significantly improved at D84 for the 0.18% HA group only ( + 2.5 ± 6.2 s for the sum of three measurements, p = 0.01) – Table 2. The increase was at the limit of significance for the 0.30% HA group ( + 1.68 ± 6.83, p = 0.08). However, the difference between both groups was not statistically significant. The Schirmer test increase was only significant for the 0.30% HA group at D35 versus baseline ( + 2.14 ± 7.77 mm/ 5 min, p = 0.040). The difference between the groups was also not significant neither at D35 nor D84. After 84 days of treatment, the Oxford score still significantly improved for both groups (−41.9% with 0.30% HA, p < 0.001 and −34.8% with 0.18% HA, p < 0.001), and the inter-group difference remained non-significant. Conjunctival staining by lissamine green and evaluation with van Bijsterveld score showed a significant improvement of van Bijsterveld score with 0.30% HA at both D35 and D84 compared to D0 (−0.69 ± 1.11 at D35 and −0.97 ± 1.73 at D84, p < 0.001) and a significant improvement with 0.18% HA at D84 only (−0.68 ± 1.32, p = 0.010).
Summary of performance results comparing 0.30% HA to 0.18% HA in patients with moderate to severe dry eye disease.

p < 0.001 compared to baseline.
HA: hyaluronic acid; TBUT: tear break-up time.
After 35 and 84 days, the score of ocular symptoms significantly improved compared to D0 for the 0.18% HA and the 0.30% HA groups (−11.11 ± 13.75 with 0.30% HA and −10.50 ± 19.39 with 0.18% HA on D35; −17.77 ± 16.52 with 0.30% HA and −15.33 ± 23.60 0.18% HA on D84; p < 0.001 at all follow-ups). Intergroup difference was not statistically significant. Patients’ evaluation of the DEQ-5 questionnaire also showed a significant improvement for both HA concentrations throughout the follow-up. At D84, DEQ-5 decreased of −5.82 ± 4.09 compared to baseline for the 0.30% HA group (p < 0.001) and of −4.77 ± 5.32 for the 0.18% HA group (p < 0.001). The difference between the 2 groups was not statistically significant.
Treatment satisfaction and global product performance were evaluated (Figure 2). On D35, most of the investigators and the patients were satisfied or very satisfied with the global performance of the treatments: 65.7% of the investigators and 77.2% of the patients for 0.30% HA versus 82.9% and 68.6%, respectively, for 0.18% HA. The results were even more pronounced at D84, and a greater satisfaction was noted for both treatments: 76.4% of the investigators and 82.4% of the patients for 0.30% HA versus 82.4% and 76.5%, respectively, for 0.18% HA. There was no significant difference between both concentrations whatever the time point.
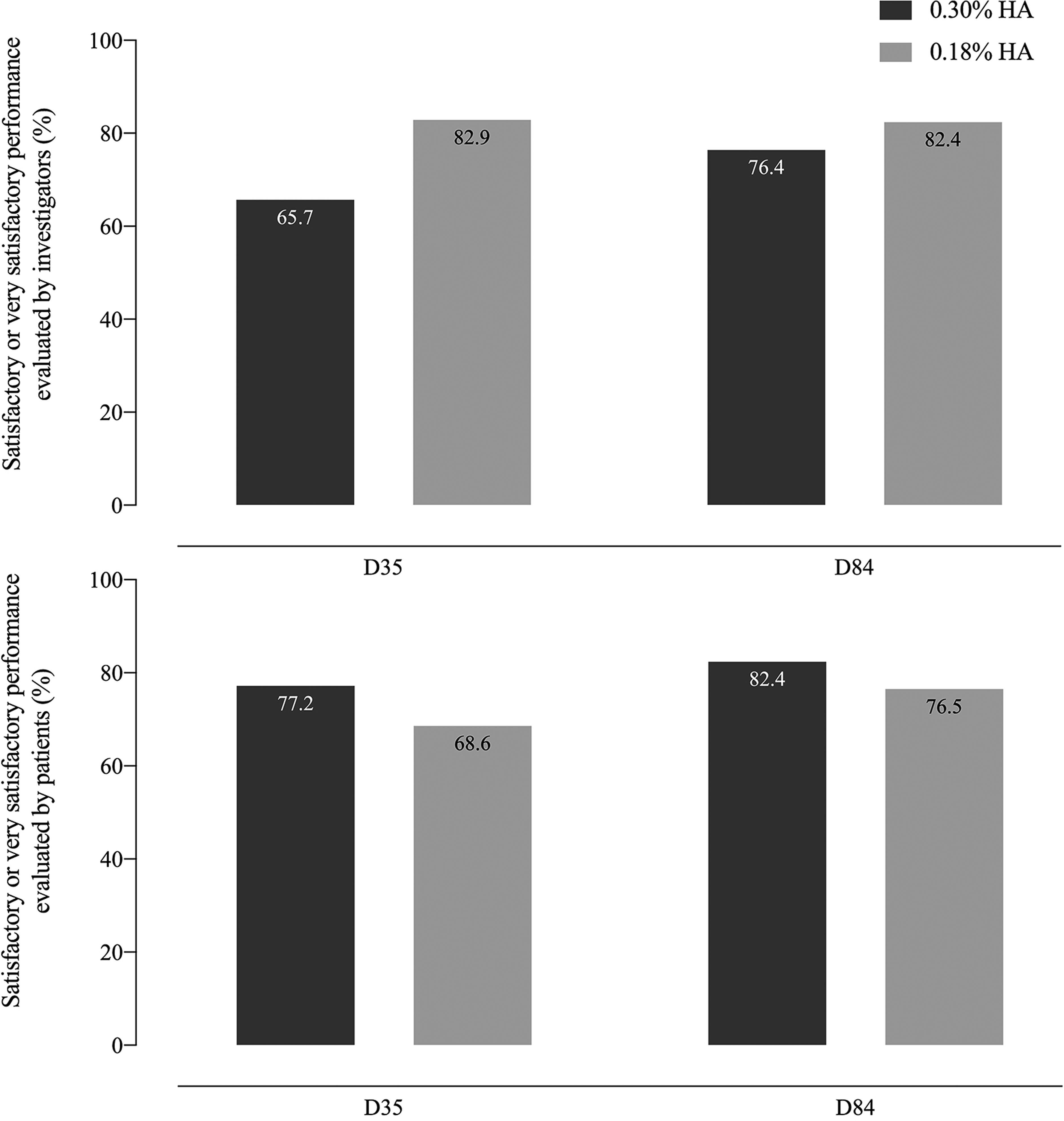
Global performance of 0.30% and 0.18% hyaluronic acid (HA) evaluated by investigators and patients with moderate to severe dry eye disease over 84 days of follow-up. The figures represent the percentage of subjects “satisfied” or “very satisfied” with either concentration.
The frequency of use of the products was similar for all patients. At D84, drops were instilled 4, 5 and 6 times per day in 55.9%, 23.5% and 17.6% of the 0.18% HA group versus 76.5%, 8.8% and 11.8% of the 0.30% HA group, respectively (p = 0.272). Similar results were also obtained at D35 (p = 0.516).
Regarding safety, both products were globally well tolerated. Three patients of each group had at least one mild or moderate adverse event related or possibly related to the study product or the study method. No patient reported severe adverse events. In the 0.30% HA group: one patient reported mild blurred vision; one patient reported mild stinging and mild scintillating scotoma possibly related to the study methods (adverse events reported during the wash-out period, before product use); and one patient reported moderate blurred vision, sensory disturbance, photophobia, and ocular hyperemia. In the 0.18% HA group: one patient reported mild eye pain; another patient reported mild eye burns; and a third patient reported moderate eyelid pruritus, all possibly related to the study product.
Discussion
DED is a disorder of the lacrimal functional unit due to tear deficiency and/or excessive tear evaporation and is associated with symptoms of ocular discomfort, dryness, burning, soreness and/or grittiness. The condition also features other clinical signs, including ocular surface damage (superficial keratitis), tear film instability, decreased tearing and tear hyperosmolarity. Current management of ocular dryness is largely addressed by the suppression of contributing factors (e.g. medications) and prescription of a wide range of artificial tears (eyedrops, fluid solutions, or ophthalmic gels).12,13 Even though no one product is considered best for everyone, 14 treatment can be optimized by adopting a “patient-centric” strategy that combines products of different viscosities and/or complementary components. In vitro experiments have shown that HA was significantly better than carboxymethylcellulose and hydroxypropyl methylcellulose in retaining water and protecting corneal epithelial cells from dehydration. 15 Several authors have therefore reported the beneficial effects of HA, in different concentration, on tear film stability and ocular surface integrity.5,6,16–18
Impaired tear film stability is a fundamental factor in diagnosing DED and is routinely assessed in clinical practice by measuring TBUT. 19 Many studies have reported improved corneal wettability and lengthened TBUT with the use of HA, highlighting its water-retention and lubrication properties.5,7,16,20,21 In our study, TBUT was equally improved in all groups, showcasing an improvement in tear quality. Schirmer test results, on the other hand, were only significant for the 0.30% HA group at D35. Even though these results are to be interpreted carefully because of normal baseline Schirmer measurements, they were still in favour of the higher 0.30% HA concentration at D35 with its subsequent higher water retention property.
Corneal fluorescein Oxford score is assumed to be an important diagnostic tool for corneal damage, reflecting corneal epithelium integrity. You et al. 7 compared 3 concentrations of HA in experimental DED on mice and reported a significantly lower corneal fluorescein staining score in the higher concentration 0.30% HA group compared to the 0.10% and 0.18% groups from day 21 to day 28 of follow-up. Our study on humans shows comparable corneal and conjunctival staining by fluorescein for 0.18% and 0.30% HA at D35 and D84. These results are in good agreement with previous clinical data from a phase IV clinical trial 20 that showed that the effects of 0.10%, 0.15% and 0.30% HA were comparable in improving the health of the ocular surface and stimulating corneal epithelium healing. In addition, the lissamine green staining is important in reflecting conjunctival epithelium integrity. Unlike Park et al. 20 who reported no significant intergroup difference, our results showed an earlier improvement of the van Bijsterveld score for the 0.30% HA group at D35. These results suggest that the application of higher concentration HA eye drops could have a beneficial and faster effect on the conjunctival epithelium healing and is therefore better adapted for more severe DED with extensive epithelial damage. Studies with longer follow-up and bigger enrolment are needed to confirm this theory.
Gel and ointments often cause more blurred vision than solutions and are normally reserved for bedtime use. 14 Noteworthy, our study did not find any statistical difference between the two HA concentrations in terms of DEQ-5 score or treatment satisfaction (for both patients and physicians) suggesting that daily use of 0.30% HA, as of 0.18% HA, does not cause gel-related blurred vision disturbances when prescribed for severe DED patients. Frequency of instillation was similar between the 2 groups, but this result may have been biased by the study protocol (frequency of instillation ranging between 4 and 6 times per day).
Because of its improved rheology and viscoelasticity that contribute to its long-lasting properties, the 0.30% HA gel lasts longer on the surface of the eye providing prolonged wettability and improved comfort for the patient without particularly causing blurriness or decreasing overall satisfaction. In fact, in their investigation on five marketed tear substitutes, Paugh et al. 22 found that precorneal residence time partly mirrored the viscosity, being more than a two-fold greater than saline for more viscous formulations. Therefore, daily use of 0.18% HA drops along with evening/bedtime instillation of 0.30% HA gel can be used for moderate DED. Daily use of 0.30% HA is possible for more severe cases, particularly if they initially present with poor or blurry vision (such as in Gougerot-Sjögren patients or post-surgery).
Both concentrations were well tolerated, and all adverse events were resolved by the end of the study.
Our study has some limitations. First, the duration of the study is short and may be insufficient to evaluate the long-term impact of different concentrations of HA on DED. Second, due to the strict inclusion criteria, the sample population was relatively small. Another limitation would be the absence of a placebo group. Finally, included patients must have had increased ocular staining with only one of the Schirmer test or TBUT altered. Because of the different included DED etiologies, some patients had normal or high measurements of Schirmer which may have biased the overall baseline average. Results concerning this endpoint are therefore to be interpreted carefully and might limit their generalizability. Even though the study is limited in size and duration, it has the advantage of being conducted on humans instead of murine dry eye models and of being multicentric, randomized and investigator blinded.
In conclusion, our study demonstrates the non-inferiority of 0.30% HA compared to 0.18% HA over an 84-day follow-up period, in patients with moderate to severe DED. Both concentrations induce significant and equivalent improvement in DED signs and symptoms, with reduced Oxford and van Bijsterveld scores and less ocular dryness symptoms. A non-significant numerical decrease in the frequency of instillation and faster conjunctival epithelial healing were noted for the 0.30% HA, probably related to its gel structure and higher HA concentration providing prolonged comfort without causing visual disturbances. Based on these findings, its use might be of interest particularly at bedtime or during the day in more severe conditions.
Footnotes
Acknowledgements
The authors thank Dr Emilio Dorronzoro Ramírez at Hospital Universitario La Moraleja, Madrid, Spain; Dr Naon Kim at Hospital Torrejon de Ardoz, Madrid, Spain; Dr David Salom at Hospital de Manises, Valencia, Spain; and Dr Victor Charoenrook at Instituto-Centro de oftalmología Barraquer, Barcelona, Spain for their valuable contribution.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Pr. Calonge, Pr. Baillif, Pr. Gain, Dr. Paw, Dr. Mearza and Pr. Cochener have no conflict of interest. Dr. Sahyoun reports working as a Medical Advisor for Horus Pharma at the time of publication.
Funding
This study was supported by Horus Pharma, Saint-Laurent-du-Var, France.