
Abstract
Objectives
Feline coronavirus (FCoV) is widely acknowledged to gain pathogenicity within the host to cause the lethal disease feline infectious peritonitis (FIP). Most FIP cases are caused by viruses in genotype 1 (FCoV-1) via an ‘internal mutation’ in the spike gene. Genotype 2 (FCoV-2) has risen to prominence based on the emergence of FCoV-23, a highly pathogenic novel variant from Cyprus that has a deletion in the N-terminus (domain 0) of spike. In this study, we conducted a retrospective analysis of three cases of FCoV-2 in the USA: two (cats 344 and 346) from 2013 and one (cat 597) from 2016. Cats 344 and 597 exhibited clinical signs consistent with FCoV infection for at least 2 months. The third cat (cat 346), the daughter of cat 344, was euthanized shortly after showing signs.
Methods
We collected a total of 20 tissue, fluid and fecal samples from the three cats. We characterized the FCoV-2 in these samples using whole-genome sequencing, genetic and phylogenetic analyses, and viral RNA quantification.
Results
Whole-genome sequencing revealed that the two cats exhibiting long-term signs of FCoV infection (cats 344 and 597) each had a distinct deletion in domain 0 of spike, which was present in all examined tissues. Cat 346, the daughter of cat 344, who displayed signs for only a short period, had an intact (or long) spike gene. Low RNA titers of the FCoV-2 with the short version of spike were detected in the feces of cats 344 and 597 (2.52–5.28 RNA copies/μl).
Conclusions and relevance
Our data are consistent with a model whereby FCoV-2 displaying the long version of spike is transmitted between cats, whereas the short version is generated within each cat after a prolonged infection and spreads rapidly throughout the body. We suggest that the high pathogenicity of FCoV-2 is associated with an ‘internal deletion’ in the spike gene.
Keywords
Introduction
Feline and canine coronavirus (FCoV and CCoV, respectively) are alphacoronaviruses highly prevalent worldwide. FCoV and CCoV are classified into two genotypes, types 1 and 2, with extensive recombination occurring between them. 1 FCoV type 2 (FCoV-2) is a genotype resulting from the recombination between FCoV type 1 (FCoV-1) and CCoV type 2 (CCoV-2). 2 Both FCoV-1 and FCoV-2 are found in two biotypes: a generally apathogenic feline enteric coronavirus (FECV) and a highly pathogenic, macrophage-tropic form known as feline infectious peritonitis virus (FIPV). 3 Infection with the highly pathogenic form leads to feline infectious peritonitis (FIP), a disease that is usually fatal without antiviral treatment. 4 FCoV-1 is more widespread among domestic cats worldwide, whereas various recombinant variants of FCoV-2 have mostly been reported in small outbreaks.5,6 In 2023, a large-scale outbreak of a novel FCoV-2-like recombinant variant (known as FCoV-23) swept through the free-ranging cat population in the Republic of Cyprus. 7 The prevalence remains unclear, with estimates in the range of 10,000 7 to 300,000 8 cases of FIP. One isolated case of FCoV-23 has been introduced into the UK through travel, 9 and the spread of this highly pathogenic FCoV poses a threat to global cat populations, warranting surveillance of this and similar emerging viruses in other countries.
The spike (S) protein of coronaviruses plays a crucial role in pathogenicity by facilitating host-cell entry via receptor binding (S1 domain) and membrane fusion (S2 domain). For FCoV-1, pathogenicity has been linked to mutations generated within the cat, 10 with key mutation(s) now thought to lie within the spike gene. 11 FCoV-1 has a cleavage site between S1 and S2 (the S1/S2 cleavage site), which primes the virus for fusion. 12 In transmissible, low-pathogenicity FCoVs, this cleavage site is characterized by a consensus motif (SRRSRR|S), where R is arginine, S is serine and | indicates the cleavage site. Point mutations disrupting this motif and reducing the proteolytic priming of spike are strongly linked to pathogenicity. 11 Other markers of pathogenicity for FCoV-1 include a mutation in a residue located in the S2 domain (known as M1058L), 13 although its role remains unclear. The ‘internal mutation’ hypothesis suggests that the accumulation of these (and possibly other) mutations enables the virus to gain macrophage tropism. 10 FCoV-2 and FCoV-23 lack the S1/S2 cleavage site and the ‘1058’ residue, and there is less understanding of the molecular mechanisms underlying the emergence of pathogenic variants.14,15
Two characteristics define the spike of FCoV-23: (1) it likely originated from a ‘pantropic’ CCoV-2 (pCCoV-2) known to circulate in the Mediterranean basin, with pathogenic CCoV-2 NA-09 detected in Greece in 2009 being its closest relative; 16 and (2) two versions of spike have been identified: a long one and a short one. 7 The short version has a deletion in the N-terminus of the spike protein that encompasses most of domain 0. This deletion has been reported in approximately 90% of FCoV-23/FIP cats and varies in size among individuals (152–245 amino acids). 7 To date, deletions in domain 0 have not been reported in other FCoV-2s (although only seven complete FCoV-2 spike sequences are currently available; see Table S1 in the supplementary material). The deletion contributes to the pathogenicity of this novel FCoV by enhancing fusogenicity and accelerating viral cell entry in feline and canine tissues. 17
In this study, we conducted a retrospective analysis of three cases of FCoV-2 in the USA: two cases (cats 344 and 346) from 2013 and one (cat 597) from 2016. Using whole-genome sequencing, phylogenetic analyses and RNA quantification, we detected and characterized FCoV-2 with both long and short spike-gene variants. Altogether, the results in this study are consistent with a model whereby FCoV-2 displaying the long version of spike is transmitted between cats, while the short version is generated within each cat after a prolonged infection and spreads rapidly throughout the body. We refer to this as the ‘internal deletion’ hypothesis. This study is the first report of FCoV-2 displaying short versions of spike in the USA and indicates that this mechanism is not exclusive to the evolution of FCoV-23.
Materials and methods
Cornell University’s Institutional Animal Care and Use Committee reviewed and approved the study procedures (approval number 2012-0116).
Sample collection and processing
Tissue, feces and ascites were collected in 2013 from cats 344 and 346 in Plainview, AR, and in 2016 from cat 597 in Philadelphia, PA. Cat 344 was a 3-year-old British Shorthair (BSH) female residing in a multi-cat environment with five other cats. In early March 2013, she started showing signs of ascites and was euthanized on 23 May 2013. Cat 346 was a 9-month-old BSH female and the daughter of cat 344, which started showing signs of ascites on 6 May 2013 and was euthanized on 4 June 2013. Cat 597 was an 11-month-old domestic shorthair male residing in a multi-cat environment. The cat had a history of upper respiratory infections and diarrhea. On 15 March 2016, he started showing signs of ascites and was euthanized on 5 May 2016. Histological tissue processing and detection of FCoV by immunohistochemistry using the FIPV3-70 antibody were carried out at the Cornell University College of Veterinary Medicine Animal Health Diagnostic Center. Slides were scanned using the Aperio CS2 (Leica Biosystems) image capture device (Cornell University College of Veterinary Medicine) and visualized using the Aperio ImageScope – Pathology Slide Viewing Software 12.4.6 (Leica Biosystems). After collection, samples were frozen at –80°C until further use.
FCoV screening, RNA quantification and whole-genome sequencing
The samples used in this study were selected following an initial screening conducted immediately after sample collection. This screening targets two regions in the spike gene of FCoV-1, as previously described. 18 Samples that test negative in these FCoV-1-specific PCRs are flagged as possible FCoV-2 and stored until further use. RNA was extracted from the 20 collected samples (Table 1), as described previously.19,20 FCoV RNA was detected using the TaqMan Fast Virus 1-Step Master Mix for qPCR (Thermo Fisher Scientific) and previously published primers/probe. 21 Whole genomes of FCoV were sequenced using hybridization capture with a panel targeting FCoV-1, FCoV-2, CCoV-1 and CCoV-2 (design ID: TE-92694309; Twist Biosciences). The synthesis of dsDNA, library preparation, hybridization capture and next-generation sequencing were performed as previously described. 19 Paired-end reads were trimmed for quality using Trimmomatic. 22 Trimmed reads were mapped against 115 sequences of FCoV-1, FCoV-2, FCoV-23, CCoV-2, CCoV-1 and transmissible gastroenteritis virus (TGEV) (see Table S2 in the supplementary material) using BWA-MEM2. 23 Quality trimming and mapping were carried out in Galaxy V22.01. 24 Viral RNA was quantified using the QX200 droplet digital PCR (ddPCR) System, the 1-Step RT-ddPCR Advanced Kit for Probes (Biorad), and the primers and probes shown in Table S3 in the supplementary material.
Samples collected and results from the screening and sequencing of feline coronavirus (FCoV) from cats 344, 346 and 597

This qPCR assay does not differentiate between FCoV-1 and FCoV-2
Neg = negative; Pos = positive; qPCR = quantitative PCR
Genetic analyses
The whole-genome sequences obtained in this study (20 from FCoV-2 and one from FCoV-1) were aligned with 115 additional whole genomes of selected FCoV-1, FCoV-2, FCoV-23, CCoV-1, CCoV-2 and TGEV (see Table S2 in the supplementary material) using the MUSCLE algorithm. 25 Percentage similarity between sequences was calculated in Geneious Prime 2023.2.1 (Biomatters), recombination breakpoints were detected in the Recombination Detection Program (RDP) 5 26 using the RDP method 27 and similarity plots were generated in SimPlot 5.1. 28 The complete open reading frame (ORF)1a region (11,613 nt) was aligned for the three sequences detected in this study and selected FCoV-23, FCoV-2, FCoV-1 and CCoV-2 sequences (see Table S2 in the supplementary material). This alignment was used to construct a maximum likelihood (ML) phylogeny using MEGA 12.0.11. 29 The amino acid sequence of the spike protein of the three sequences obtained in this study and selected FCoV-23, FCoV-2 and CCoV-2 (see Table S2 in the supplementary material) were aligned using the Clustal Omega algorithm. 30 Excluding domain 0, the alignment of the spike protein was used to construct an ML phylogeny using MEGA 12.0.11. 29 All phylogenies were visualized and color-edited using iTOL v7.2. 31 Both the alignment of the complete spike protein and the one excluding domain 0 were used to generate a similarity plot using SimPlot 5.1. 28 The accession numbers for all the sequences used in each analysis are listed in Table S2 in the supplementary material.
Results
The 20 samples collected from the three cats tested positive for FCoV by quantitative PCR (Table 1). Whole-genome sequencing, recombination analyses and similarity plots revealed that all the FCoVs detected were recombinants between FCoV-1 and CCoV-2 (ie, FCoV-2) (Figure 1). The recombinant region of the FCoV-2 detected in cats 344 and 346 (FCoV-2/344 and FCoV-2/346, respectively) covered most of the ORF1b and 3abc genes (RDP, P <0.001) and the one from cat 597 (FCoV-2/597) included only the spike gene (RDP, P <0.001). In the fecal sample of cat 344, co-infection with FCoV-1 (FCoV-1/344.6) (Table 1) was detected. The S1/S2 cleavage site of FCoV-1/344.6 was SRRSRR|S, and residue ‘1058’ was M, consistent with the shedding of a low-pathogenicity phenotype. The FCoV-2s detected in all the tissues of each cat were nearly identical (>99.9% similarity); therefore, only one sequence for each FCoV-2 will be used as a reference from this point forward. A maximum of three nucleotide differences was observed between the whole genomes of FCoV-2 from the same cat, and these were randomly distributed across ORFs 1a and 1b.
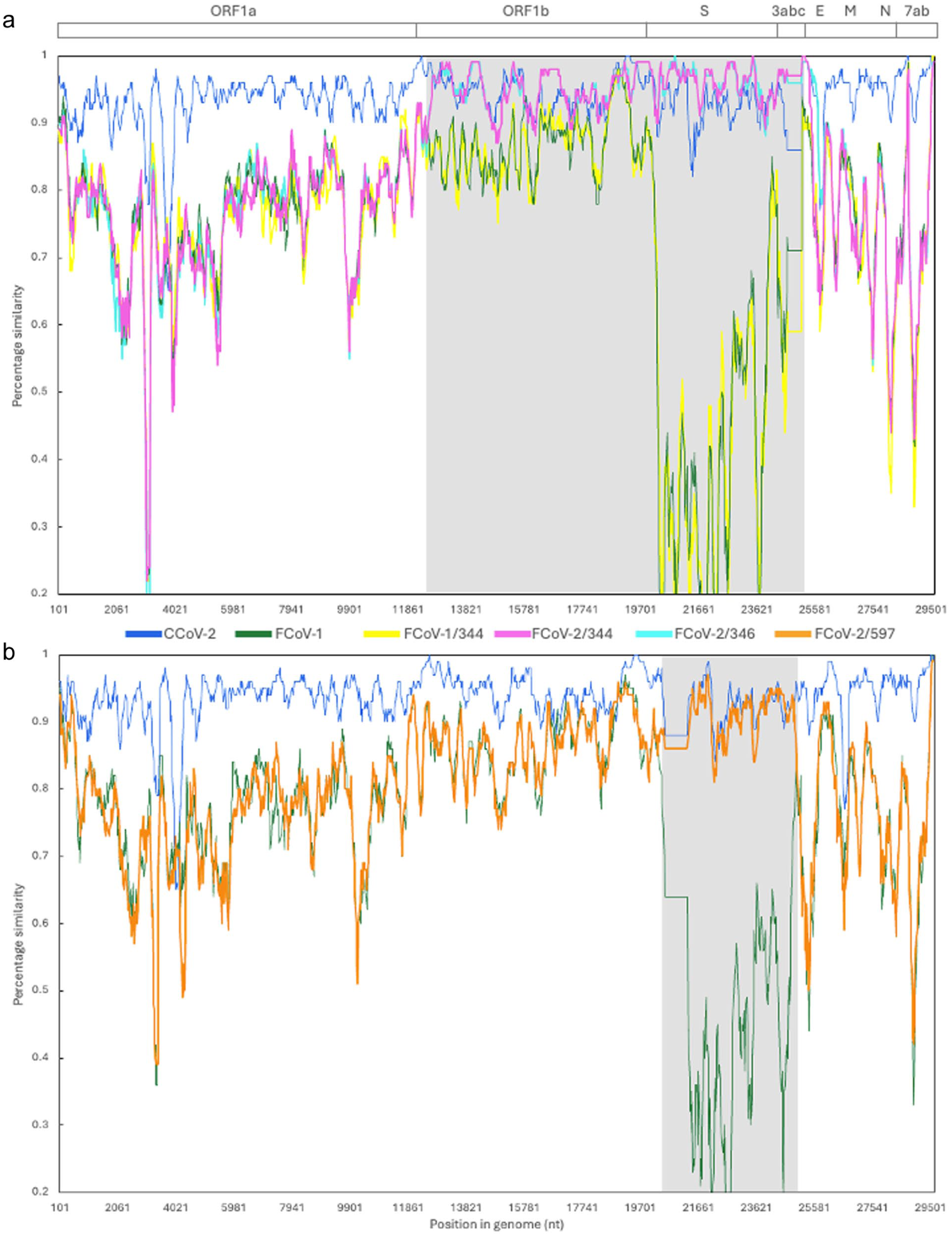
Similarity plots of the whole-genome sequences of (a) feline coronavirus (FCoV)-1/344, FCoV-2/344 and FCoV-2/346 and (b) FCoV-2/597. At the top is a graphical depiction of the general organization of the genome of FCoV/canine coronavirus (CCoV), including the non-structural (open reading frame [ORF]1a, ORF1b), structural (S, E, M, N), and accessory (3a, b, c and 7a, b) genes. In each graph, the nucleotide similarity across the entire genome (~29,500 nt) is compared between a reference sequence (CCoV-2 CB-05/Italy/20025/KP981644) and the sequences obtained in this study (FCoV-1/344 in yellow, FCoV-2/344 in fuchsia, FCoV-2/346 in cyan, FCoV-2/597 in orange), FCoV-1 (in green) and CCoV-2 (in blue). Recombinant regions are shaded in gray and include the ORF1b, spike and 3abc genes for FCoV-2/344 and FCoV-2/346, and the spike gene for FCoV-2/597. In these regions, the detected FCoV-2s are more similar to CCoV-2 (in blue) than to FCoV-1 (in green). FCoV-1/344 is more similar to FCoV-1 than the other sequences throughout the whole genome. The plots were generated using the Kimura 2-parameter distance model, with a 250 bp window and a 40 bp step. Sequences used in the plot: FCoV-1 RM/USA/2012/JQ404410, CCoV-2 TN449/USA/2012/JQ404410, FCoV-2 WSU79-1683/USA/1979/JN634064
Phylogenetic analysis of ORF1a (Figure 2) showed that FCoV-1/344, FCoV-2/344 and FCoV-2/346 form a single group, suggesting a common origin for these FCoVs. Genetic analysis of the spike gene of the three FCoV-2s detected showed that FCoV-2/344 has a deletion of 459 nt (153 amino acids) and FCoV-2/597 has a deletion of 636 nt (212 amino acids) (Figure 3). Each deletion was detected in all the tissues processed from each individual (Table 1). No deletion was detected in the spike of FCoV-2/346 (Figure 3). No co-infection with the long and short versions was detected in any of the processed tissues. When compared with FCoV-23 (Table 2), major deletions were also detected in ORF3b (30 nt) and ORF3c (170 nt) of FCoV-2/344, ORF3b (30 nt) of FCoV-2/346 and ORF3c (29 nt) of FCoV-2/597 (Table 2). ORF3a of FCoV-3/344 and FCoV-2/346 were predicted to be longer (35 nt). Excluding the deletions identified in the various FCoV-2 variants, the genomes of FCoV-2/344 and FCoV-2/346 exhibited a similarity of 97.4%. Excluding the recombinant region, FCoV-1/344 and FCoV-2/344 were 94.1% similar.

Maximum likelihood phylogenetic tree of open reading frame (ORF)1a (11,613 nt) of feline coronavirus (FCoV) and canine coronavirus (CCoV). The FCoVs sequenced in this study (344, 346, 597) are in bold. FCoV-1 is in green, FCoV-2 is in blue and FCoV-23 is in purple. Circles in the branches indicate bootstrap percentage values for 1000 replicates. The size of each circle is proportional to the bootstrap value; the scale is displayed at the bottom left. Best-fitting nucleotide substitution model: GTR+G+I
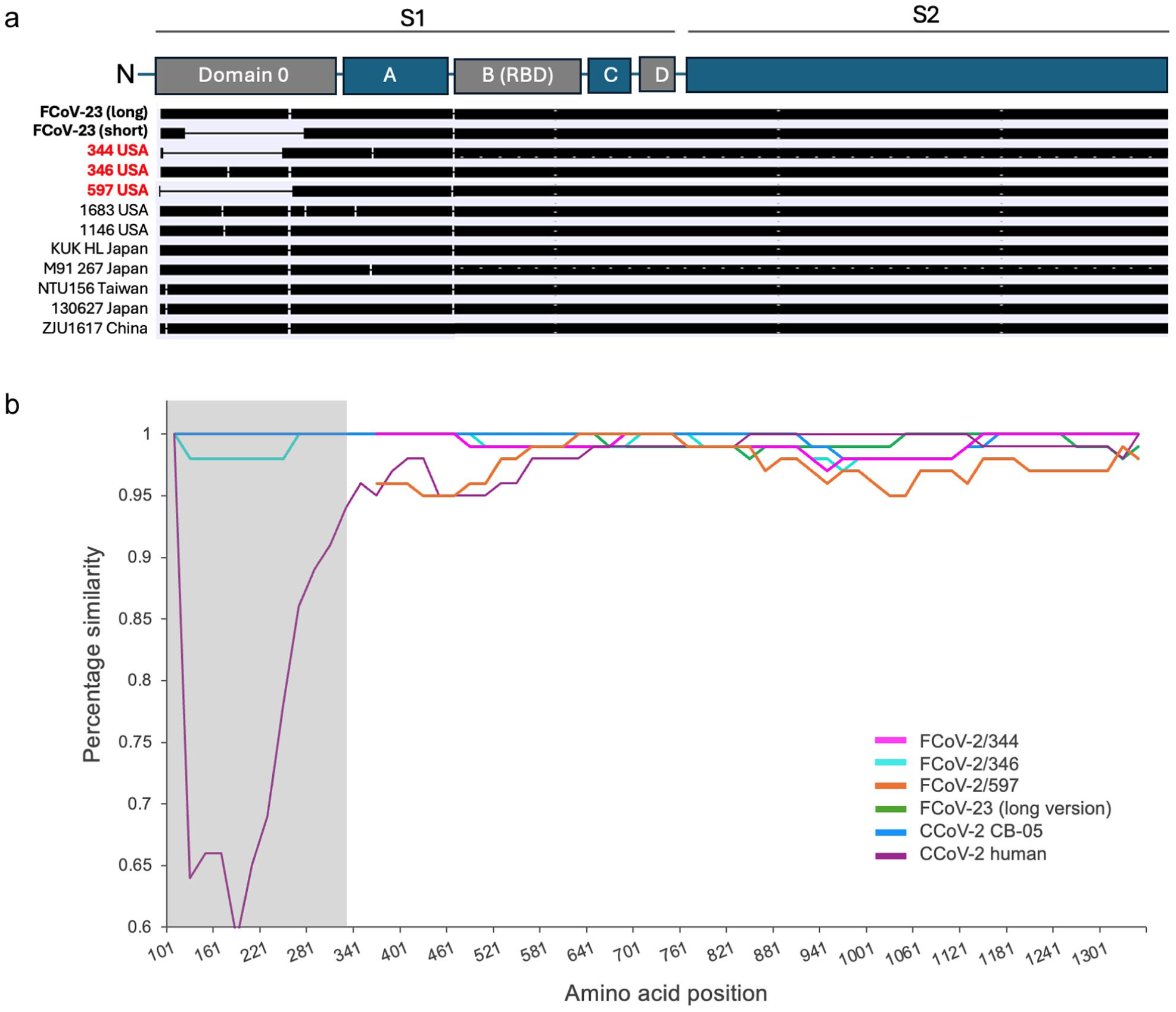
Genetic diversity of the spike protein of feline coronavirus (FCoV)-2, FCoV-23 and canine coronavirus (CCoV-2). At the top is a graphical depiction of the different domains of the spike protein. (a) Graphical representation of the different sizes of the spike protein from different FCoV-2 and FCoV-23 variants. Each horizontal black bar represents the length of the spike protein of 12 FCoVs, including the three identified in this study (in red) and the long and short versions of FCoV-23 (in bold). Deletions are shown as thin horizontal lines and are all in domain 0. The short version of FCoV-23, FCoV-2/344 and FCoV-2/597 have deletions of different sizes. FCoV-2/346 does not have a deletion in this region. (b) Similarity plot of the amino acid sequence of the spike protein (1341 amino acids). CCoV-2 NA09/2009/Greece/JF682842, which is the closest variant to FCoV-23, was used as a reference. Domain 0 is shaded in gray in accordance with the placement shown in the graphical depiction of spike at the top of the figure. FCoV-2/344 and FCoV/346 feature deletions in this region; thus, comparisons in this area are not possible. The plot was generated using the Kimura 2-parameter distance model, with a 200 bp window and a 20 bp step. Sequences used in the plot: CCoV-2 CB-05: KP981644, CCoV-2 human: MZ420153, FCoV-23 (long version): PQ133182. RBD = receptor binding domain
Size (in amino acids) of the seven open reading frames (ORFs) in which major deletions have been detected in feline coronavirus (FCoV)-2 and FCoV-23

A similarity plot of the amino acid sequence of spike, using the closest variant to FCoV-23 as a reference (CCoV-2 NA09/2009/Greece), showed that FCoV-2/344, FCoV-2/346, FCoV-2/597, FCoV-23 and pCCoV-2 CB-05/2005/Italy exhibit over 90% amino acid similarity across the spike protein sequence, even when including domain 0 for FCoV-2/346 and the long version of FCoV-23 (Figure 3). In contrast, domain 0 of the CCoV-2 detected in humans is highly dissimilar (Figure 3). Phylogenetic analysis of the amino acid sequence of spike, excluding the deletion region, showed that the three FCoV-2 variants reported in this study form a distinct group, closely related to a group that includes FCoV-23 and other pCCoV-2 variants reported in Europe (Figure 4).

Maximum likelihood phylogenetic tree of the amino acid sequence of the spike protein (1081 amino acids) of feline coronavirus (FCoV) and canine coronavirus (CCoV). The domain 0 region (where large deletions occur) was excluded. CCoV-2 and FCoV-2 detected in the USA between 2012 and 2016, including the three reported in this study (in bold), are in orange. Pantropic CCoVs reported in Europe are in green. FCoV-23 is in purple. Circles in the branches indicate bootstrap percentage values for 1000 replicates. The size of each circle is proportional to the bootstrap value, and the scale is shown at the top left. Best-fitting nucleotide substitution model: GTR+G+I
FCoV-2 RNA levels varied among the 20 samples tested from the three cats (Figure 5). For cat 597, FCoV-2 RNA levels ranged from 2.5 RNA copies/μl in feces to 93,174.5 RNA copies/μl in the mesenteric lymph node (see Table S4 in the supplementary material). For cat 344, the concentration of FCoV-2 RNA ranged from 5.3 RNA copies/μl in feces to 449,125 RNA copies/μl in the omentum. The quantification of FCoV-1 in the fecal sample from cat 344 revealed an RNA load of 22,543.5 RNA copies/μl (see Table S4 in the supplementary material). For cat 346, FCoV-2 RNA levels were 47.7 RNA copies/μl in ascites and 434.5 RNA copies/μl in spleen (see Table S4 in the supplementary material). The immunohistochemical detection of FCoV-2 revealed extensive viral infection in tissues from cats 344 (Figure 6) and 597 (Figure 7) when compared with the spleen sample of cat 346 (Figure 6).

Quantification of feline coronavirus (FCoV)-2 and FCoV-1 RNA in tissues, fluids and feces of cats 344 (fuchsia), 346 (cyan) and 597 (orange). FCoV-1 RNA was detected and quantified only in the feces of cat 344 (bar with diagonal lines). Bars with squares represent the concentration of FCoV-2 RNA quantified in the feces of cats 344 and 597. The values shown are from two replicates
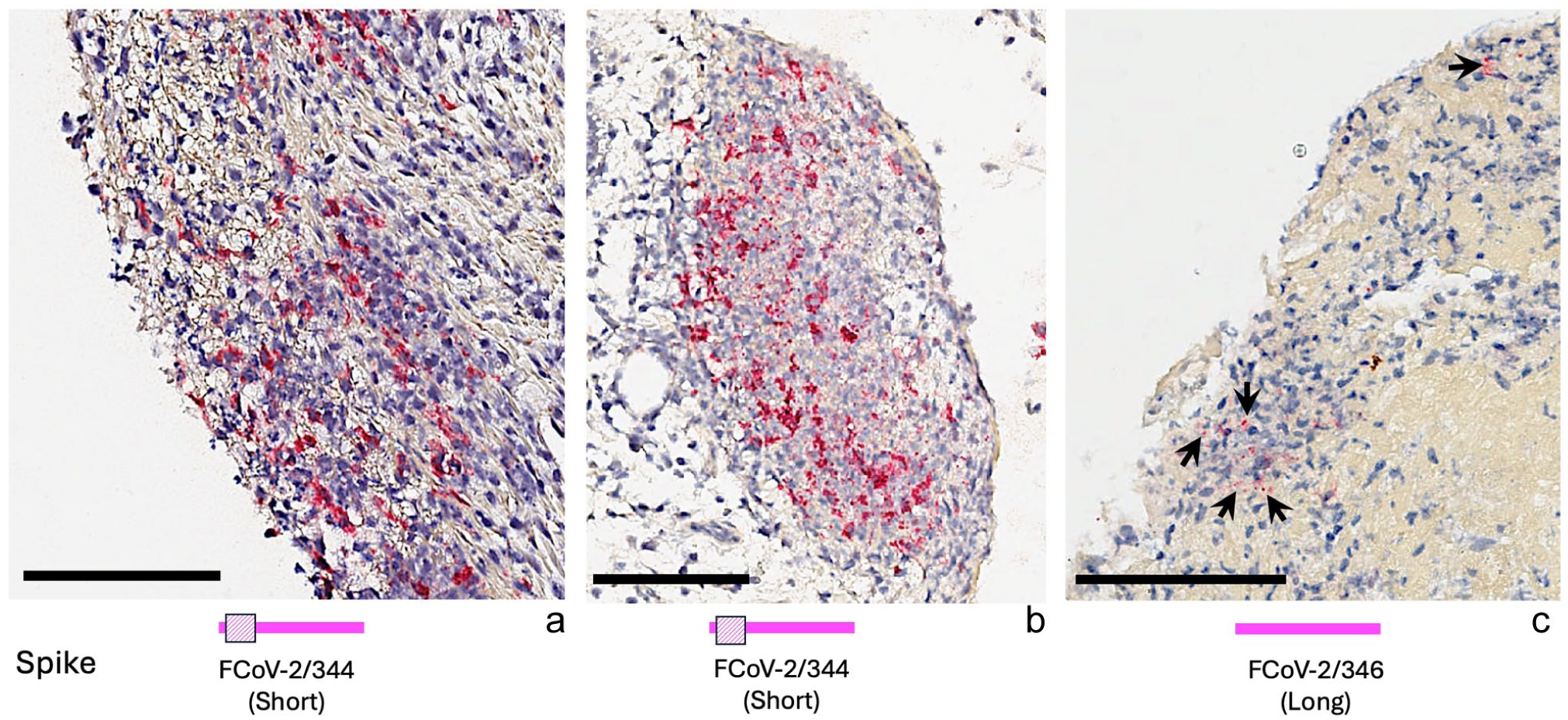
Detection of feline coronavirus (FCoV)-2 (in burgundy) in tissues of cats 344 and 346 by immunohistochemistry. Below each picture is a cartoon illustrating the length of the spike protein: FCoV-2/344 has a short spike, and FCoV-2/346 has a long spike. FCoV-2 in (a) the small intestine and (b) the omentum of cat 344, and (c) the spleen of cat 346 (arrows). Black bars in the lower left corner of each photograph indicate a 100 μm scale

Detection of feline coronavirus (FCoV)-2 (in burgundy) in tissues of cat 597 by immunohistochemistry. In all tissues, FCoV-2/597 displayed a short spike protein, as shown in the bottom left of the figure. FCoV-2 in (a) the large intestine, (b) the liver, (c) the spleen and (d) the mesenteric lymph node of cat 597. Black bars in the lower left corner of each photograph indicate a 100 μm scale
Discussion
FCoV-23 is a highly pathogenic FCoV that emerged in Cyprus, resulting in a widespread, deadly outbreak among free-ranging cats. 7 FCoV-23 resembles FCoV-2 as it is a recombinant of FCoV-1 and CCoV-2, which most likely exchanged the spike gene with a pCCoV-2. The spike protein of FCoV-23 is characterized by two variants that differ in length. The shorter variant contains a variable-size deletion within domain 0 (Table 2). Variants displaying the deletion exhibit enhanced fusogenicity and cell entry, suggesting that the deletion is a significant pathogenicity factor. 17 We conducted a molecular retrospective survey of FCoV-2 detected in three cats in the USA in 2013 and 2016, revealing two variants of FCoV-2 with different recombination breakpoints (Figure 1) and deletions in domain 0 of spike (Figure 3). We detected different deletions in domain 0 of the spike protein in the FCoV-2 found in cats 344 and 597 (Figure 3). The deletion size differed between individuals (153 amino acids in FCoV-2/344 and 212 amino acids in FCoV-2/597) (Figure 3), but it was identical within each cat and was identified in all tissues and fluids examined (Table 1). We did not detect deletions in the spike of the FCoV-2 from cat 346, the daughter of cat 344 (Figure 3). Although the duration of infection for all cats is unknown, we hypothesize that cats 344 and 597 had prolonged infections because their medical histories indicate that they exhibited clinical signs of FCoV-2 infection (ascites, diarrhea) for at least 2 months. Persistent FCoV infection in cats has been widely reported32,33 and most cats with chronic diarrhea are infected with FCoV. 34 Cat 346 was euthanized approximately 1 month after the initial development of ascites. Therefore, we assume that this cat was infected for a shorter duration than cat 344, which had ascites for nearly 3 months (from early March to late May 2013). Our results suggest that prolonged infection with FCoV-2 leads to deletions in domain 0 of spike and that viruses with these deletions rapidly spread within each host, causing extensive infection (Figures 6 and 7). Analogous to the ‘internal mutation’ hypothesis of FCoV-1, 10 we call this the ‘internal deletion’ hypothesis.
The transmission of FCoV-2 primarily occurs through the fecal–oral route, and the viral load in feces is crucial for viral transmission. For example, 4/5 (80%) cats experimentally inoculated with feces containing a low FCoV RNA concentration did not seroconvert or shed FCoV in their feces. 35 In contrast, 17/17 (100%) of those inoculated with feces containing a high viral RNA concentration rapidly seroconverted and began shedding the virus in their feces. 35 Cats that are persistently infected and shed high titers of FCoV RNA in their feces are the most significant source of FCoV in multi-cat settings. 32 Therefore, measuring viral RNA titer in feces can serve as a proxy for the likelihood of FCoV transmission. Cats 344 and 346 were living in the same house, and the FCoV-2s detected in these cats were 97.4% similar, had the same recombination breakpoints (Figure 1) and were grouped together in the phylogenies of ORF1a (Figure 2) and spike (Figure 4). The main difference between them was that FCoV-2/346 has the long variant of spike, while FCoV-2/344 has the short variant (Figure 3). Although we detected FCoV-2 RNA with the short version in the feces of cats 344 and 597, the viral RNA load was very low (2.5 RNA copies/μl for FCoV-2/597 and 5.3 RNA copies/μl for FCoV-2/344) (see Table S4 in the supplementary material) compared with the viral RNA load in the tissues and fluids (range 828 ± 0.2 to 449,125.0 ± 1438.9 RNA copies/μl for FCoV-2/344, and 9.8 ± 1.3 to 93,174.5 ± 1167.4 RNA copies/μl for FCoV-2/597) (see Table S4 in the supplementary material) and the RNA load of the FCoV-1 detected in the feces of cat 344 (22,543.5 ± 118.1) (Figure 4). These results suggest that the FCoV-2 with the long variant of spike was transmitted between cats 344 and 346, and the deletion observed in FCoV-2/344 developed within the host and spread systemically in cat 344 (Table 1). Supporting this and the ‘internal deletion’ hypothesis is the fact that 90% of the cats infected with FCoV-23 in Cyprus also exhibit deletions that vary in size and are specific to each individual. 7 In the case of the Cyprus outbreak, high FCoV-23 RNA loads have been reported in feces; however, it remains unclear whether the long or the short variant of spike is being shed. 7
To determine whether individuals with prolonged infections develop deletions in domain 0, it is essential to monitor the evolution of the spike gene during FCoV-2 outbreaks. In an outbreak in Taiwan, the duration of FCoV-2 infection, measured from the onset of fever to death, was in the range of 15–67 days in four cats. 5 Although the spike gene was sequenced in some of these cats, only the 3’-end was targeted; thus, the evolution of the 5’-end, where domain 0 is located, among cats with different infection times remains unknown. Phylogenetic and genetic comparisons of the entire spike protein sequence revealed that the FCoV-2s reported in this study are closely related to pCCoV-2s and FCoV-23 (Figure 4). In addition, their spike protein is highly conserved, displaying over 90% amino acid similarity across the entire protein (Figure 3), including domain 0, which is highly divergent in other coronaviruses, such as CCoV-2 reported in humans (Figure 3). Considering the close genetic relationship with pCCoV-2 and FCoV-23, and the detection of pathogenic variants with domain 0 deletions in the USA, it is crucial to start surveillance programs for FCoV in multi-cat households such as shelters, as well as free-ranging domestic cats in the USA, to prevent a large-scale outbreak similar to the one in Cyprus.
Our results suggest that rapid molecular detection and characterization of FCoV are essential for preventing the development and spread of highly pathogenic viruses with deletions in the spike gene. In the feces of cat 344, we detected co-infection with FCoV-1 and FCoV-2. The viral RNA load of FCoV-1/344 in feces was higher than that of FCoV-2/344 (see Table S4 in the supplementary material); however, this FCoV-1 lacked any of the pathogenicity markers in spike (ie, no mutations were identified in the S1/S2 cleavage site or in residue ‘1058’). In this case, the pathogenic variant that spread systemically and caused extensive infection was an FCoV-2 displaying a deletion in spike. Therefore, detection methods targeting only the spike of FCoV-1 can detect FCoV-1 without markers of pathogenicity while overlooking co-infection with highly pathogenic FCoV-2 variants. Likewise, detection methods that focus on regions outside the spike gene, which are more conserved, would have also missed the co-infection, as FCoV-2/344 and FCoV-1/344 were highly similar (94.1% pairwise similarity, excluding the recombinant region). We recommend using the hybridization method used in this study, which allowed us to detect both co-infection with FCoV-1 and FCoV-2, as well as variants of FCoV-2 with major deletions not only in spike but also in other genes (Table 2). To date, hybridization capture has not been widely applied in disease surveillance. 36 In part, these limitations in application are due to the costs and centralized facilities needed for this technology; however, we consider this technology to be ideally suited to the diverse recombinant genomes present in the viral species alphacoronavirus-1 (Alphacoronavirus suis), which includes feline and canine coronaviruses. 37 This study alerts the companion animal community to the risks and scope of infection by the viral species alphacoronavirus-1 and informs the development of future technology with clinical application in low-resource settings, such as veterinary practices, animal shelters and field situations. 38 Future work will focus on deploying new technologies to inform studies of the highly complex disease syndrome caused by canine and feline coronaviruses.
Conclusions
We propose an internal deletion model for within-host generation of highly pathogenic FCoV-2 through the loss of spike domain 0 (Figure 3). We further propose that FCoV-23 is not in itself a novel FCoV-2 variant but is a highly impactful presentation of this mechanism based on the high population density of free-roaming cats on the island of Cyprus.
Supplemental Material
Table S1
Size of spike, in nucleotides and amino acids for available FCoV-2 variants.
Supplemental Material
Table S2
Sequences used in the different analyses and phylogenies of this study.
Supplemental Material
Table S3
Primers and probes used for quantifying the RNA of FCoV-1 and FCoV-2.
Supplemental Material
Table S4
Viral RNA quantification in the samples collected from three cats.
Footnotes
Acknowledgements
We thank Dr Mandi de Mestre for allowing us to use the ddPCR system to quantify viral RNA and Donald Miller for teaching us how to use it.
Author note
All the sequences obtained in this study were deposited in GenBank under accession numbers PX306341–PX306360 and PX132896.
Supplementary material
The following files are available as supplementary material:
Table S1: Size of spike, in nucleotides and amino acids for available FCoV-2 variants.
Table S2: Sequences used in the different analyses and phylogenies of this study.
Table S3: Primers and probes used for quantifying the RNA of FCoV-1 and FCoV-2.
Table S4: Viral RNA quantification in the samples collected from three cats.
Conflict of interest
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This research was funded by the Morris Animal Foundation (grants D25FE-713 and D25FE-706) and Cornell Feline Health Center.
Ethical approval
The work described in this manuscript involved the use of non-experimental (owned or unowned) animals. Established internationally recognized high standards (‘best practice’) of veterinary clinical care for the individual patient were always followed and/or this work involved the use of cadavers. Ethical approval from a committee was therefore not specifically required for publication in JFMS. Although not required, where ethical approval was still obtained, it is stated in the manuscript.
Informed consent
Informed consent (verbal or written) was obtained from the owner or legal custodian of all animals described in this work (experimental or non-experimental animals, including cadavers, tissues and samples) for all procedures undertaken (prospective or retrospective studies). No animals or people are identifiable within this publication, and therefore additional informed consent was not required.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.