
Abstract
Objectives
The aim of this study was to determine the pharmacokinetics of furosemide in cats following intravenous (IV), oral and transdermal administration.
Methods
This study used six healthy adult cats in a three-phase design to compare plasma furosemide concentrations in cats that received one IV 2 mg/kg dose of furosemide, one oral 2 mg/kg dose of furosemide and 3 days of q12h dosing with 2 mg/kg furosemide transdermally applied to the ear pinna.
Results
After IV administration the elimination half-life was (mean and coefficient of variation) 2.25 h (72%), systemic clearance was 149 ml/kg/h (27.4%) and volume of distribution was 227 ml/kg (22%). After oral administration the terminal half-life was 1.2 h (18.7%), peak concentration was 3.4 μg/ml (51.7%) and bioavailability was 48.4%. The transdermal plasma concentrations were undetectable or very low at most time points, and pharmacokinetics were not determined from the transdermal dose.
Conclusions and relevance
Furosemide was rapidly eliminated in cats after oral and IV administration and is probably best administered orally at least q12h in cats with heart failure. The oral dose absorbed was approximately 50%, but the absorption from transdermal administration was negligible.
Introduction
Furosemide is the most common diuretic drug used in veterinary medicine. It is routinely administered intravenously or intramuscularly in acute heart failure, and orally for chronic control of volume overload associated with cardiac disease in dogs and cats.1,2 It is the most important medication administered to cats in congestive heart failure (CHF) and it is generally necessary to administer at least daily in cats for long-term control once CHF has developed. 2
Because some owners have difficulty administering chronic oral medications to feline patients, some have used compounded transdermal furosemide in cats. In addition to being easier for the owners to administer, transdermal administration of medications is often less stressful for the owner, as well as the cat, which would likely improve compliance and disease control. Various studies evaluating a transdermal approach for various medications, including phenobarbital, 3 amlodipine, 4 buspirone 5 and methimazole,6–9 and many others (reviewed in Davidson and Papich), have been performed. 10 These studies show that, except for a limited few drugs, transdermal administration is not appropriate for cats because of low and inconsistent absorption, and/or irritation and dermatitis to the pinna. Because furosemide has some properties that may make it suitable for transdermal absorption (meets the ‘rule-of-five criteria’), this study was undertaken to determine if the transdermal approach might be an acceptable option in some feline patients. This study was conducted with a crossover design to compare intravenous (IV), oral and transdermal administration at a dose of 2 mg/kg. The transdermal formulation was applied repeatedly to determine if repeated administration would improve absorption. Additionally, because the pharmacokinetics of furosemide have not previously been published in cats, systemic availability following a single oral dose was also examined.
Materials and methods
This study was approved by the Animal Care and Use Committee at the University of Pennsylvania Veterinary School. Six adult (1–3 years of age) cats weighing 2.0–3.7 kg were used in a crossover design study. A 22 G catheter was placed for blood draws during each phase of the study and a 24 G IV catheter was placed for administration of furosemide during the IV phase of the study. In the first phase of the study, each cat received oral furosemide (2 mg/kg) once. In the second phase of the study, each cat received IV furosemide (2 mg/kg) once and in the final phase of the study, transdermal furosemide (2 mg/kg) was administered q12h for 3 days in each cat. A washout period of at least 3 weeks was ensured between each of the experiments.
Transdermal furosemide was compounded in a lipoderm base (PCCA Lipoderm) and was used within 7 days of production. Potency of the transdermal compound was tested at two concentrations (40 mg/ml and 80 mg/ml) 3 and 7 days after production. The measured potencies ranged from 97–102%. The drug was applied to the contralateral ear, morning and night. Blood samples were obtained immediately prior to dosing and at 0.5, 1, 2, 3, 4, 6 and 8 h post-administration of furosemide (IV and oral phase) and at 1, 2, 3, 4, 6 and 72 h (during the transdermal phase). Cats had free access to water during sample collection. One cc of blood was drawn into a syringe with heparin-saline and re-administered to the cat after obtaining 1 cc for the blood sample. The blood sample was transferred to lithium heparin tubes and centrifuged within 30 mins of collection. The plasma was transferred to freezer tubes and stored at −70°C until analyzed.
Plasma drug analysis
Furosemide in plasma was analyzed by reversed-phase high-pressure liquid chromatography using a method that was identical to the assay recently used for canine studies in our laboratory.11,12 To validate the assay for this study in cats, blank (control) feline plasma was fortified with furosemide at three concentrations (high, medium and low) and measured to ensure that concentrations fell within the acceptance criteria for the assay. All calibration samples were made from fortified feline blank plasma. Blank samples (time zero collected prior to drug administration) from each cat were analyzed to measure background noise and verify that there were no interfering peaks in the window of interest in the chromatogram.
Pharmacokinetic analysis
Furosemide plasma concentrations were plotted on a semi-logarithmic graph to examine the shape of the curve for preliminary selection of an appropriate model.For the IV dose, a two-compartment pharmacokinetic model was fitted to these data using a computer program (Phoenix WinNonlin version 7.0; Certara). The model is described by the following Equation 1.

C is the plasma concentration, A is the distribution phase y-axis intercept, e is the base of the natural logarithm, t is time after injection, alpha (α) is the distribution rate constant, B is the elimination phase y-axis intercept and beta (β) is the elimination phase rate constant (terminal phase). Secondary parameters calculated include distribution (α) and elimination (β) half-lives (T½), micro-distribution rate constants, area under the curve (AUC), apparent volume of distribution at steady-state, systemic clearance and mean residence time.
For the oral dose, a one-compartment model, with first-order oral absorption was fitted to the data using the following Equation 2.
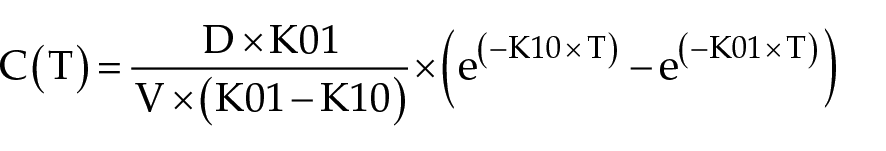
C is the plasma concentration, T is time, K01 is the rate of oral absorption, K10 is the first-order elimination rate taken from the terminal slope, D is the dose and V is the volume of distribution. Secondary parameters from the model included the peak concentration (CMAX), area under the concentration vs time profile (AUC), and the respective half-lives (T ½ ) for each rate. For each route of administration, descriptive statistics were generated (mean, SD and coefficient of variation [%]). Oral bioavailability (F) was calculated from the AUC ratios (AUCORAL/AUCIV). Because this was a crossover study, each cat served as its own control in calculating F.
The selection of the correct pharmacokinetic model was verified by examining goodness-of-fit criteria, predicted vs observed plots and plots of weighted residuals vs time plots. Because the concentrations from the transdermal application were low or undetected, pharmacokinetic analysis could not be performed on the transdermal data.
Results
IV, oral and transdermal administration of furosemide (2 m/kg once [IV and PO] or 2 mg/kg q12h for 3 days [transdermal]) was well tolerated in all cats. No changes were noted in physical examination or behavior during the study period. IV furosemide resulted in a shorter elimination half-life (T½) than orally administered furosemide; however, furosemide administration by either route resulted in a short T½ in the plasma (<1.2–2.3 h). Plasma concentration vs time profiles of furosemide after the three routes of administration are presented in Figures 1 and 2. Values for the plasma pharmacokinetic parameters for furosemide administration after oral and IV routes of administration are presented in Tables 1 and 2. Because of low or undetectable concentrations in most samples after transdermal administration, pharmacokinetic analysis was not performed on these data. The concentrations are shown in Figure 2.

Plasma concentrations in cats after oral (blue circle) and intravenous (IV; red diamond) administration of furosemide at a dose of 2 mg/kg by each route. Each point represents the mean ± SD of six cats, with the solid line representing the fitted line derived from pharmacokinetic modeling and data in Tables 1 and 2. Transdermal points are not shown because they are near zero

Plasma concentrations in cats after oral (blue circle) and intravenous (IV; red diamond) administration of furosemide at a dose of 2 mg/kg by each route. Each point represents the mean ± SD of six cats, with a solid line for oral and IV representing the fitted line derived from pharmacokinetic modeling and data in Tables 1 and 2. The x-axis is extended to 72 h (with a break at 10–70 h) and the y-axis is converted to logarithmic scale to show transdermal concentrations (black triangle symbol)
Pharmacokinetic data from intravenous administration at a dose of 2 mg/kg

A and alpha are the intercept and rate of the distribution phase, respectively, with corresponding half-life (T½); B and beta are the intercept and rate of the elimination phase, respectively, with corresponding half-life (T½)
AUC = area under the curve; CL = systemic clearance; K10, K12 and K21 = micro-distribution rate constants; MRT = mean residence time; Vc = apparent volume of distribution of the central compartment; Vss = apparent volume of distribution at steady-state; CV = coefficient of variation
Pharmacokinetic data from oral administration at a dose of 2 mg/kg

T½ = half-life; CMAX = peak (maximum) plasma concentration; TMAX = time to peak concentration; AUC = area under the curve; TLAG = lag time for oral absorption; F = fraction of oral dose absorbed, expressed as a percentage; CV = coefficient of variation
Discussion
The IV and oral plasma furosemide declined rapidly with a mean T½ of 2.25 h (IV) and 1.2 h (oral) (Table 1). Furosemide action derives from secretion across the renal tubule from the plasma to the lumen of the nephron. Therefore, these short half-lives should correspond to a short duration of action. The clearance of furosemide measured in these healthy cats (Table 1) is in the same range as creatinine clearance, supporting the observation that furosemide clearance in cats is entirely dependent on renal clearance. The apparent volume of distribution of furosemide in these cats was approximately equivalent to extracellular water volume.
We did not perform any pharmacodynamic studies in these cats after furosemide administration. A study in dogs demonstrated that the diuretic response was decreased above a plasma concentration of 10 µg/kg and the half maximum diuretic effect was obtained at a plasma concentration of 1.5 µg/ml, similar to a reported level of 1 µg/ml in humans. 13 If these concentrations have similar effects in cats, the duration of diuretic activity can be estimated from examining Figures 1 and 2. In dogs and humans, the diuretic response induced by furosemide was maintained for 3 h after IV administration. 13 The short T½ in this study in cats suggests that dosing at least q12h is likely to result in superior diuresis than q24h dosing.
Plasma furosemide levels were not detectable or very low after transdermal administration, even though it was applied for multiple doses. Seventy-two hours after starting transdermal administration, plasma furosemide concentration remained very low (0.07 µg/ml ± 0.06 µg/ml) (Figure 2) but was detectable in all six cats. This suggests that there may be some accumulation of drug over time. This accumulation after multiple doses notwithstanding, it appears that administration of transdermal furosemide in the vehicle and dose used for this study will not be effective and is not recommended.
Without further study, an explanation for poor transdermal absorption in these cats from this formulation is undetermined. Proposed characteristics for an ideal transdermal drug include size <500 d, melting point of <200°C, the saturated aqueous solution should be in a relatively physiologic range (pH 5–9) and the delivered dose should be <15 mg/day. 14 Additionally, a drug that is either too lipophilic or hydrophilic may not be suitable. 14 Furosemide also meets the ‘rule-of-five’ criteria that suggests good transdermal absorption. 15 Based on these criteria, furosemide has at least some characteristics that suggest it may be successful by the transdermal approach. Furosemide has a melting point of 206°C, a molecular weight of 330.74 and a total daily dose of 15 mg/kg is rarely necessary in cats. However, it is only slightly soluble in water and, instead, soluble in acetone or methanol (http://www.chemicalbook.com/ChemicalProductProperty_EN_CB2445739.htm).
The vehicle used in the present study contains a proprietary liposomal component that is stable upon refrigeration and has greater resiliency in the presence of ionic substances (PCCA Lipoderm). This vehicle was used because of previous success with other drugs in cats. However, because the vehicle is proprietary, not all of its characteristics are available for review. Further work is necessary to optimize the vehicle used for furosemide to maximize penetration of furosemide by a transdermal approach in future studies. Although this study involved a small group of cats, each cat served as its own control when comparing the three routes of administration.
Conclusions
Oral absorption of furosemide was approximately 50% (mean), but transdermal absorption was negligible. With the current evidence, transdermal furosemide administration appears unlikely to be effective in cats with CHF. Because of the short T½ and examination of Figures 1 and 2 for IV and oral administration, furosemide is probably best administered at least q12h to maintain efficacy in cats with heart failure.