
Abstract
An 11-year-old girl presented with a soft tissue lesion on the dorsal aspect of the left middle finger. Ultrasound imaging demonstrated a 2.8 cm × 0.8 cm × 0.8 cm lesion overlying the dorsal aspect of the base of the digit near the metacarpophalangeal joint. The patient’s past medical history is remarkable for neuroblastoma, diagnosed at 9 months of age, with no MYCN amplification or 1p loss. We report a pediatric schwannoma harbouring a SH3PXD2A::HTRA1 gene fusion with a distinctive serpentine histology. The lesion consisted of well-circumscribed nodules surrounded by thin EMA-positive perineural capsules. Each nodule was composed of lesional cells arranged in short fascicles with occasional clefting and a distinct “serpentine” palisading pattern. The lesion demonstrated Antoni A regions with Verocay body formation. No significant Antoni B areas were seen. The lesional Schwannian cells were bland with elongated and tapered nuclei, showing strong and diffuse positivity for S100. This pre-pubescent girl (Tanner Stage 2) is currently the youngest reported case of fusion-positive schwannoma. In addition, she has a significant prior history of a malignant neoplasm, and the lesion arose in an appendicular location.
Introduction
Schwannoma, the most common nerve sheath neoplasm, is composed almost entirely of schwannian cells.1,2 Schwannomas most commonly occur in somatic soft tissue, where they are often associated with peripheral nerves. Additional locations include those originating from spinal or cranial nerves, and those in visceral organs, especially the gastrointestinal tract, thought to arise from autonomous nerves.1,2
The understanding of schwannoma tumorigenesis has been reshaped by the recent identification of SH3PXD2A::HTRA1 gene fusion in 10% of intracranial/spinal schwannomas.
In 2016, Agnihotri et al 3 published their findings of a frequently recurrent in-frame fusion between the SH3PXD2A and HTRA1 genes in 12/125 (10%) of their schwannoma patients. The SH3PXD2A::HTRA1 gene fusion was also identified by Aaron et al, 4 who noted it in 1/14 vestibular schwannoma patients subjected to RNA sequencing. In their case series, they also found the fusion showed male predominance, occurring in 1 out of every 6 men with schwannoma. 3
Lee et al 5 have screened 215 schwannomas for their clinicopathologic characteristics and fusion status, describing that the presence of this fusion was significantly associated with serpentine histology, smaller tumors, younger patients, and peripheral somatic tissue, as seen in this case.
This pre-pubescent girl is currently the youngest reported case of fusion-positive schwannoma; the youngest patient described by Lee et al 5 is 16 years old. In addition, this case is of clinical interest as she has a significant prior history of a malignant neoplasm, and the lesion arose in an appendicular location.
Case Report
The patient is an 11-year-old with a history of neuroblastoma, diagnosed at 9 months of age. At that time, she presented with elevated urine catecholamines (HVA 39.7 and VMA 54.2 mmol/mol creatinine). Imaging revealed a lobulated heterogeneously enhancing soft tissue mass within the right thoracic inlet extending to the supraclavicular region and inferiorly to the superior posterior mediastinum (3.9 cm × 4 cm × 5 cm). There was mass effect on the superior mediastinal structures and trachea with extension to the neural foramina superiorly. There was no evidence of compression and multiple enlarged cervical lymph nodes. A bone marrow aspirate was performed, revealing neoplastic cells. The biopsy confirmed the diagnosis of neuroblastoma, Schwannian stroma poor, poorly differentiated, with favourable histology. There was no MYCN amplification or 1p loss. She achieved complete remission on completion of only 2 cycles of chemotherapy for non-high-risk neuroblastoma (ANBL0531; Doxorubicin, Cyclophosphamide, Carboplatin and Etoposide). She did not undergo radiation.
At eleven years of age, she presented with a slow-growing soft tissue mass on the dorsum of the left middle finger. The family reported that the lesion had been present for approximately 2 years. The lesion was not painful nor restricting motion. Clinical examination revealed a non-tender, mobile, and lobulated lesion to the dorsal aspect of the proximal phalanx. There were no overlying skin changes. The patient had full active and passive range of motion of the digit and no neurovascular deficits. Ultrasound imaging performed 2 years prior demonstrated a lobulated, avascular lesion measuring 1.8 cm × 0.6 cm × 0.4 cm. Up-to-date ultrasound imaging (Figure 1) demonstrated an increase in the lesion size to 2.8 cm × 0.8 cm × 0.8 cm. Surgical excision for diagnostic certainty was planned in consultation with the family.
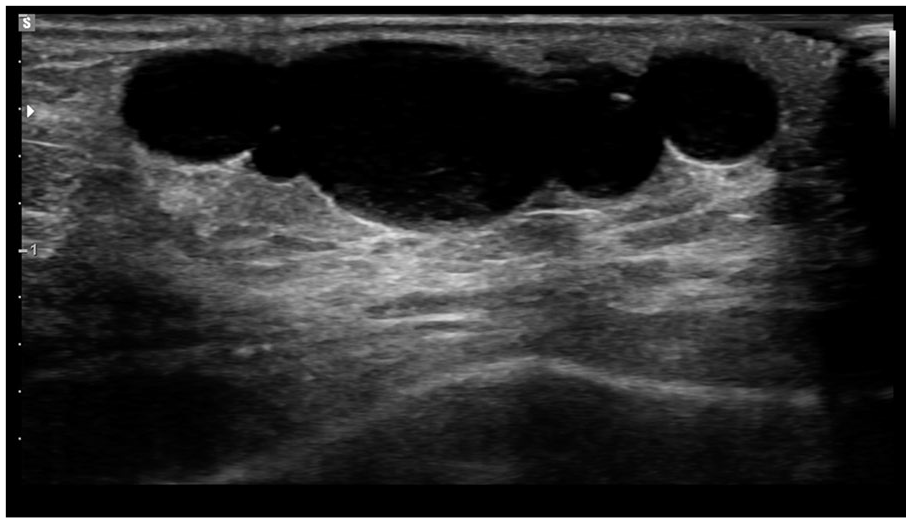
Ultrasound imaging showed a lobulated hypoechoic lesion, measuring up to 2.8 cm × 0.8 cm × 0.8 cm (CC × TR × AP), with a few thin septations and posterior acoustic enhancement overlying the dorsal aspect of the proximal third digit near the metacarpophalangeal joint. Doppler interrogation showed no internal flow.
The lesion was removed under general anesthesia and tourniquet control. A dorsal incision was made revealing a well-defined, lobulated mass lying above the extensor paratenon. The lesion was dissected easily from the surrounding tissue and sent for pathological assessment. She made an uncomplicated post-operative recovery.
Histology revealed well-circumscribed nodules surrounded by thin EMA-positive perineural capsules (Figure 2(a)). Each nodule was composed of lesional cells arranged in short fascicles with occasional clefting and a distinct “serpentine” palisading pattern. The lesion demonstrated Antoni A regions with Verocay body formation. There were no distinct Antoni B areas. The lesional Schwannian cells were bland with elongated and tapered nuclei (Figures 2(a) and (c)), showing strong and diffuse positivity for S100 (Figure 2(d)). There was no significant cytologic atypia, mitotic figures, degenerative/cystic changes, or necrosis. There were large nerve bundles in association with the lesion in the surrounding soft tissue (Figure 2(e)).
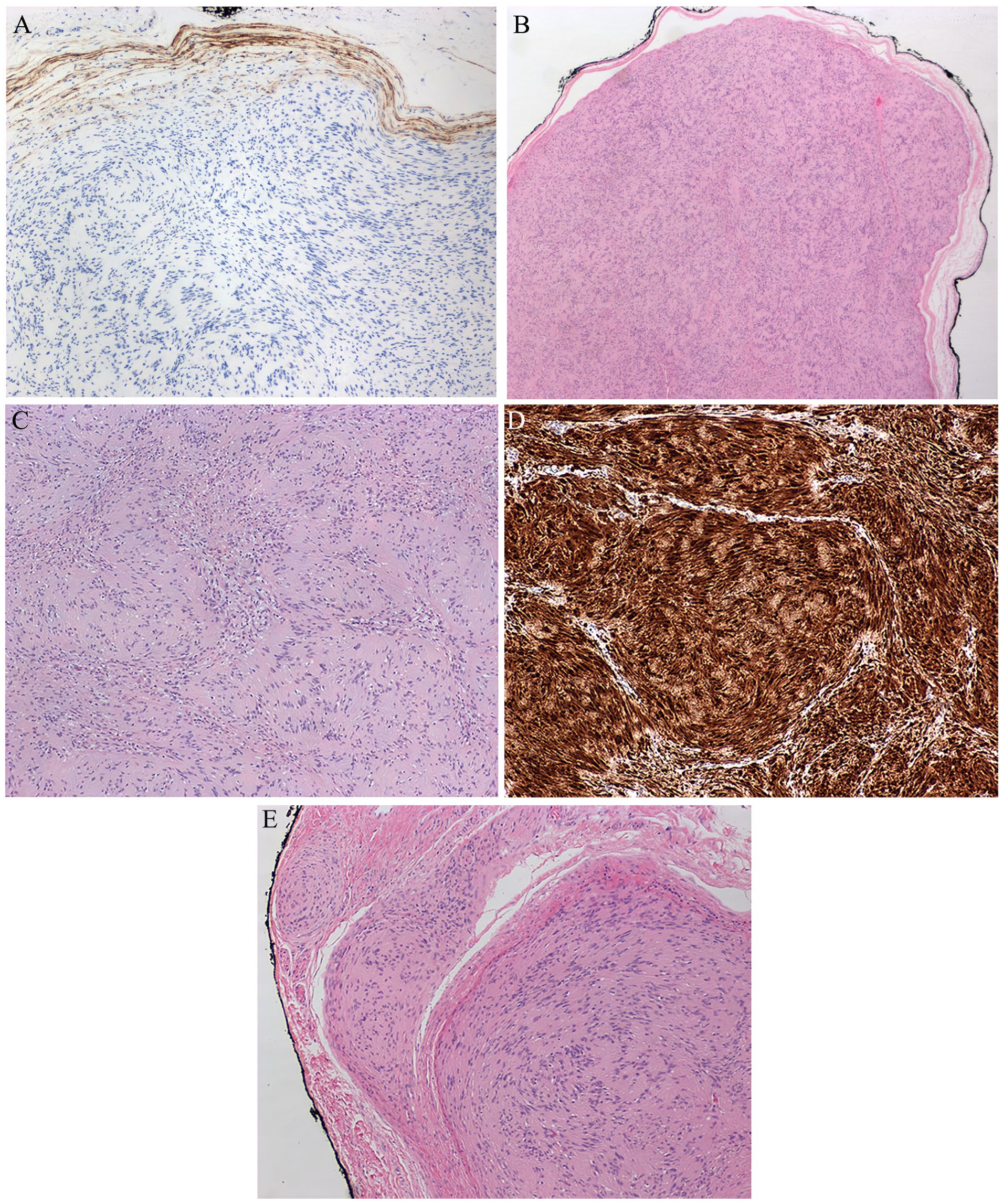
Histological and immunohistochemical features (x20). (A) Immunohistochemistry for epithelial membrane antigen (EMA) highlights perineurial cells in the capsule. (B and C) Low and high magnification views showing the distinctive distinct “serpentine” palisading pattern of the tumor; Stain: hematoxylin and eosin. (D) The cells are strongly and diffusely positive for S100. (E) Relationship of the lesion to the nascent nerves.
Illumina TruSight RNA Pan-Cancer NGS analysis was performed on paraffin-embedded tissue. RNA was extracted using the RNAstorm kit from CELLDATA for FFPE tissue. Library was sequenced on an Illumina NextSeq. The sequence was aligned to the hg19 human genomic scaffold using the Illumina STAR aligner (v2.6.1a). TruSight RNA Pan-Cancer NGS analysis identified a SH3PXD2A::HTRA1 gene fusion (Figure 3) and no single nucleotide variants through 3.89 million unique reads (sensitivity decreases in cases with fewer than 1 million unique reads).
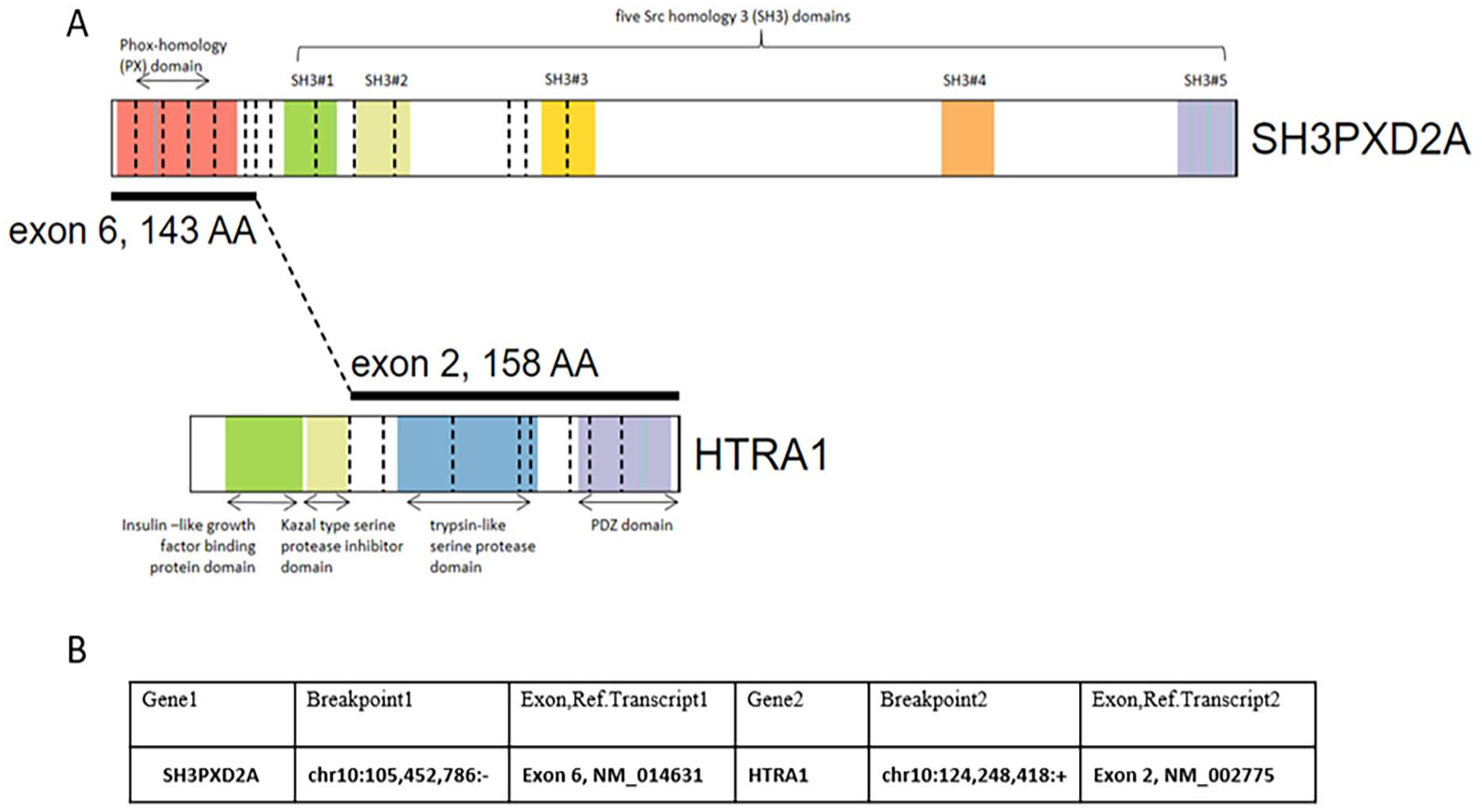
(A) Schematic showing the structure of the SH3PXD2A::HTRA1 gene fusion with the various protein domains labeled. (B) Genomic coordinates and exons involved.
Discussion
The past few decades have seen an exponential increase in molecular genetic analyses and biomarker discovery with concomitant growth of molecular pathological assays. This has led to a new age of precision or personalized medicine, allowing for more accurate diagnoses and effective treatments for malignancy. 6
A causal relationship has largely been known between schwannoma tumorigenesis and loss of expression of merlin (NF2, schwannomin), the growth inhibitory protein product of the NF2 tumor suppressor gene located at 22q12.2. 7 NF2-inactivating mutations have been detected in approximately 50–75% of sporadic cases.8-11
The SH3PXD2A::HTRA1 gene fusion has more recently been identified in roughly 10% of schwannoma cases1,3,10 and it has been reported that all of them show the distinctive “serpentine” growth pattern at least focally. It has been proposed that a serpentine growth pattern seen in 10% or more of a schwannoma is highly predictive of the fusion. 5
The expression of the SH3PXD2A::HTRA1 gene fusion results in elevated phosphorylated ERK, increased proliferation, invasion, and in vivo tumorigenesis.3,12 Genomic analysis performed by Agnihotri et al 3 demonstrated the mechanism resulting from a balanced 19-Mb chromosomal inversion on chromosome 10q.
SH3PXD2A encodes a protein that promotes invadopodia formation and regulates cell migration. 13 Invadopodia are actin-based structures that drive the proteolytic invasion of cells. SH3PXD2A protein is highly expressed in several solid tumors, including melanoma, 14 gastric, 15 breast, 16 lung, 17 oral, 18 and colon cancers. 19
HTRA1 encodes an evolutionary conserved protein quality-control factor. Recent evidence suggests that the loss of HTRA1 function causes multiple phenotypes, including the acceleration of cell growth, delayed onset of senescence, centrosome amplification, and polyploidy, suggesting an implication in cell cycle regulation. 20
Figure 3 illustrates the involved protein domains. The fusion lacks the N terminus of wild-type HTRA1 that contains the Kazal domain, an inhibitory domain for the peptidase domain of HTRA1, but gains a PXD2A domain from SH3PXD2A32. It is plausible that the lack of the Kazal domain in the fusion allows for aberrant peptidase and protease activity, whereas the PXD2A domain in the fusion protein allows for both novel interactions and sites of localization for the fusion. 3
Of note, the patient in our case had a significant clinical history of neuroblastoma. The fusion between the genes SH3PXD2A and HTRA1 is on chromosome 10, which is not among the chromosomes altered in neuroblastoma. In addition, there are no reports of the SH3PXD2A::HTRA1 fusion or any HTRA1 fusions in neuroblastoma.
Targeting of the MEK-ERK pathway was effective in Schwann cells harbouring this fusion, which suggests a possible therapeutic approach for this subset of tumors 3 and highlights the role of molecular characterization for management. However, to our knowledge, there are no clear experimental studies dissecting how the SH3PXD2A::HTRA1 gene fusion regulates the MEK-ERK signaling pathway.3,5,12
In addition, schwannomas with SH3PXD2A::HTRA1 gene fusion have shown expression of a significantly high level of HTRA1 mRNA. Further studies are needed to explore whether they could be exploitable as a biomarker. 5
In summary, we report a case of pediatric schwannoma with a SH3PXD2A::HTRA1 gene fusion. To our knowledge, this is the youngest reported case of SH3PXD2A::HTRA1 gene fusion positive schwannoma. In addition, she has a significant prior history of neuroblastoma, which has not been described in the literature in association with this lesion.
Molecular analysis of schwannomas showing this characteristic growth pattern is strongly recommended for better characterization of these tumors.
Footnotes
Authors’ Note
This work was presented in part at the 2024 Canadian Association of Pathology Annual Scientific Meeting. The manuscript has been read and approved by all the authors and the requirements for authorship in the journal instructions have been met. Each author believes that the manuscript represents honest work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical Adherence
Informed consent was obtained from the patient’s parents and all efforts have been made to uphold patient confidentiality.