
Abstract
Liver histology in infants with cystic fibrosis (CF) and persistent cholestasis is seldom reported in detail. We extend previous observation of a distinctive intrahepatic cholangiopathy (ICCF) to 3 additional infants homozygous for CFTR pathological variants and a fourth infant with a heterozygous CFTR variant, summarizing our experience in 10 infants with CFTR variants and persistent cholestasis. Cholangiograms demonstrate abnormal extrahepatic ducts in 2 infants with CF, 1 with uniform dilatation interpreted as a choledochal cyst and the other with narrow patent ducts. Liver histology in 3 CF homozygotes had prominent ductular reaction with a focally destructive cholangiolitis (inflammation of small bile ducts). The CFTR heterozygote had generalized portal edema with ductular reaction and paucity but no cholangitis. Cholestasis slowly subsided in all infants. ICCF is characterized by severe ductular reaction, prominent cholangiocyte injury, and multifocal necrotizing cholangiolitis. Local aggregates of portal ceroid might suggest previous bile leakage from damaged ducts. ICCF in liver biopsies from infants with cystic fibrosis and persistent cholestasis is unrelated to the specific CFTR genotype. Liver biopsy findings and intraoperative cholangiogram help rule out biliary atresia. ICCF is an early manifestation of CF, a likely prototype for pathogenesis of cystic fibrosis liver disease later in life.
Keywords
Introduction
Cholestatic liver disease in young infants with cystic fibrosis (CF) may mimic biliary atresia (BA), producing a diagnostic challenge. 1 Bove et al 1 reported 6 infants with persistent cholestasis and CF; 4 were interpreted as BA at laparotomy and underwent Kasai procedures but on reevaluation did not have classical biliary atresia: extrahepatic bile ducts were narrow and small gallbladders contained colorless mucin. Liver histology showed a destructive cholangiopathy that differed from typical findings in diagnostic biopsies from infants with BA where intrahepatic cholangitis is usually mild or absent.2,3 The liver biopsy findings in 10 infants, 4 new cases and 6 previously reported cases 1 support our hypothesis that the common lesion in CF infants with persistent cholestasis is a destructive intrahepatic cholangiopathy (ICCF).
Materials and Methods
Liver biopsies from 6 infants were reviewed in consultation from outside institutions and 4 were from patients at Cincinnati Children’s Hospital. Most cases were accessioned from 2010 to 2020. Histologic stains available for review included a minimum of H&E, PAS after diastase digestion, and Masson trichrome. Reticulin; CK7 IHC stains were available in more recent cases.
Special attention was directed to the presence or absence of histological features that we previously suggested helped to differentiate the intrahepatic cholangiopathy of cystic fibrosis from that of biliary atresia. Common features to both are bile duct proliferation and duct bile plugs. Differentiating features in cystic fibrosis are a degenerative cholangiopathy with foci of destructive cholangiolitis, and lack of expansile loose non-fibrotic portal stroma that is a hallmark of biliary atresia. In the current study, prevalence of portal cholangiolitis was graded from 1+ to 3+ with 1+ indicating a minority of portal areas, 2+ most portal areas, and 3+ all portal areas.
Results
Patient 1
A female infant whose brother has CF was born at term and presented with failure to thrive and jaundice early on life. The neonatal screen for CF was positive. She presented on day of life (DOL) 21 with cholestasis and failure to gain weight. Magnetic resonance cholangiopancreatography (MRCP) revealed fusiform dilatation of the hepatic duct and cystic ducts with upstream dilatation of the left and right intrahepatic bile ducts near the confluence; the gallbladder was not visualized. HIDA scan showed hepatic retention only. At laparotomy on DOL 28, the liver was firm and congested with a fine granular surface raising concerns for chronic disease. The gallbladder was small with a thick wall and resisted cannulation. Cholangiogram from the cystic duct (Figure 1(a)) showed fusiform dilatation of the common hepatic bile duct with dilation of the left main hepatic duct and tiny left hepatic duct interpreted as Type 4a choledochal cyst 4 and multiple large intrahepatic ducts with irregular lumens. The pancreatic duct was not seen. The choledochal cyst and gallbladder contained no bile and were excised to the level of portal plate. The hepatic ducts were flushed until bile was obtained. An end-to-side, retrocolic Roux-en-Y hepaticojejunostomy was performed and wedge and needle biopsies were obtained. The postoperative course was uneventful, with a short course of total parenteral nutrition (TPN).
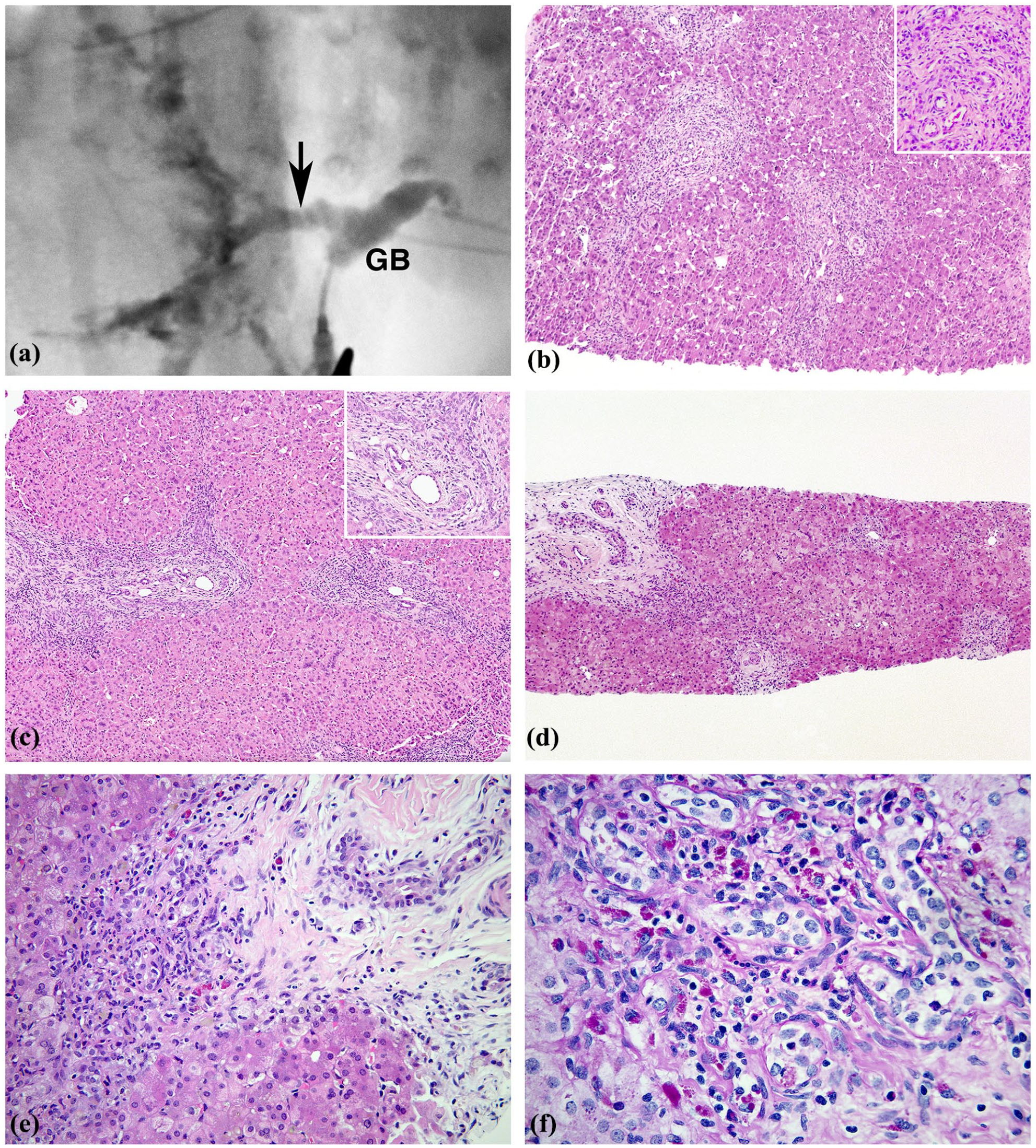
Case#1. (a) Intraoperative cholangiogram on DOL 28. Injected contrast at apex of gallbladder demonstrates normal lumen. Diffuse irregular dilatation of main hepatic duct and common bile duct interpreted as a choledochal cyst (arrow). (b) Liver DOL 28. All small portal tracts exhibit mild increase in duct profiles, mild cholangiocyte injury pattern, and minimal stromal expansion. Features of large duct obstruction are absent. H&E ×100. Inset: detail of typical portal zone. H&E ×200. (c) Liver DOL 28. A few portal zones contain proliferating ducts with acute cholangitis and pericholangitis. Portal stroma is mildly reactive. H&E ×100. Inset: Arc-like pattern of small ducts simulates ductal plate. Notable is cuboidalized cholangiocytes with nuclear variability, and mild stromal reaction. H&E ×200. (d) Liver DOL 42. Some portal zones are like those in panel B. A larger portal zone contains proliferating ducts in arc pattern lined by degenerating cholangiocytes with conspicuous zone of periportal fibrosis. H&E ×50. (e) Liver DOL 42. Area of local periportal fibrosis which shows intense necrotizing cholangiolitis, mild increase in ceroid macrophages, and regressive changes in nearby portal duct embedded in scar. H&E ×200. (f) Liver, DOL 42. Portal area with prominent bile duct proliferation features nuclear and cytoplasmic changes, pericholangitis, scattered neutrophils, and locally prominent ceroid macrophages. PAS-diastase ×200.
The choledochal cyst and gallbladder were embedded in sequence. The lining of the cyst was intact without inflammation or sclerosis and peribiliary glands were inconspicuous. The gallbladder had normal mucosa and muscularis without inflammation, excess mucus, or mucus gland hyperplasia. Intraoperative wedge liver biopsy showed severe lobular cholestasis with prominent giant cell transformation (GCT), and mild variable portal inflammation with slight reactive stromal expansion. Small bile duct profiles were mildly increased with mild neutrophilic pericholangitis (Figure 1(b) and (c)). A minority of portal tracts exhibited increased bile duct profiles in a ductal plate-like arrangement with associated cholangitis (Figure 1(c)).
Percutaneous liver biopsy on DOL 42 had persistent active cholangitis (Figure 1(d)-(f)) and focal periportal fibrosis (Figure 1(e)) now accompanied by prominent local accumulation of ceroid-rich macrophages (Figure 1(f)). The histological changes overlapped with those expected in large duct obstruction but the cholangitis, excess ceroid, and variability from one to another portal area differed from effects of large duct obstruction (e.g., BA) where uniformity is the rule.
Two pathological variants were identified in the CFTR gene: c.1521_1523del (p. Phe508del), and c.254G>A (p. Gly85Glu).
The patient had recurrent abdominal pain that was diagnosed as cholangitis and/or pancreatitis between the ages of 1 and 2 years. Portal hypertension, chronic abdominal pain attributed to recurrent pancreatitis, and both pancreatic endocrine and exocrine insufficiency were present at the age of 5 years.
Patient 2
A male infant delivered by elective cesarean section at 38 weeks gestational age (GA) weighed 2.74 kg. Imaging at 33 weeks GA had shown polyhydramnios and upper gastrointestinal obstruction with signs of meconium peritonitis. On DOL 1, a necrotic dilated duodenal segment proximal to atresia was resected. A proximal duodenostomy tube was placed for drainage and sham feeding, and a proximal ileostomy tube was placed for distal feeding. Bile was observed in the gallbladder and duodenum. Severe peritonitis restricted further exploration of the biliary system.
CF screen performed on DOL 12 reported CF related variants. The Ohio Newborn Screen reported on DOL 32 was positive for CF variants: G542X, and c.2051_2052delinsG (p. Lys684fs).
Weight gain on oral and tube feeds was slow. Bile disappeared in duodenal drainage and direct hyperbilirubinemia persisted. Serum matrix metalloproteinase 7 (MMP7) levels were slightly elevated but not suggestive of BA. On DOL 30, intestinal continuity was reestablished. The gallbladder contained clear mucus but no bile. A cholangiogram was cautiously attempted through the apex of the gallbladder but only the cystic duct was visualized. After sequential irrigations with acetylcysteine (Mucomyst), a diminutive hypoplastic extrahepatic biliary tree was visualized with flow into the duodenum but intrahepatic branches were not observed (Figure 2(a), left). Contrast injected via the cholecystotomy tube on DOL38 demonstrated a diminutive intrahepatic biliary tree (Figure 2A, right).
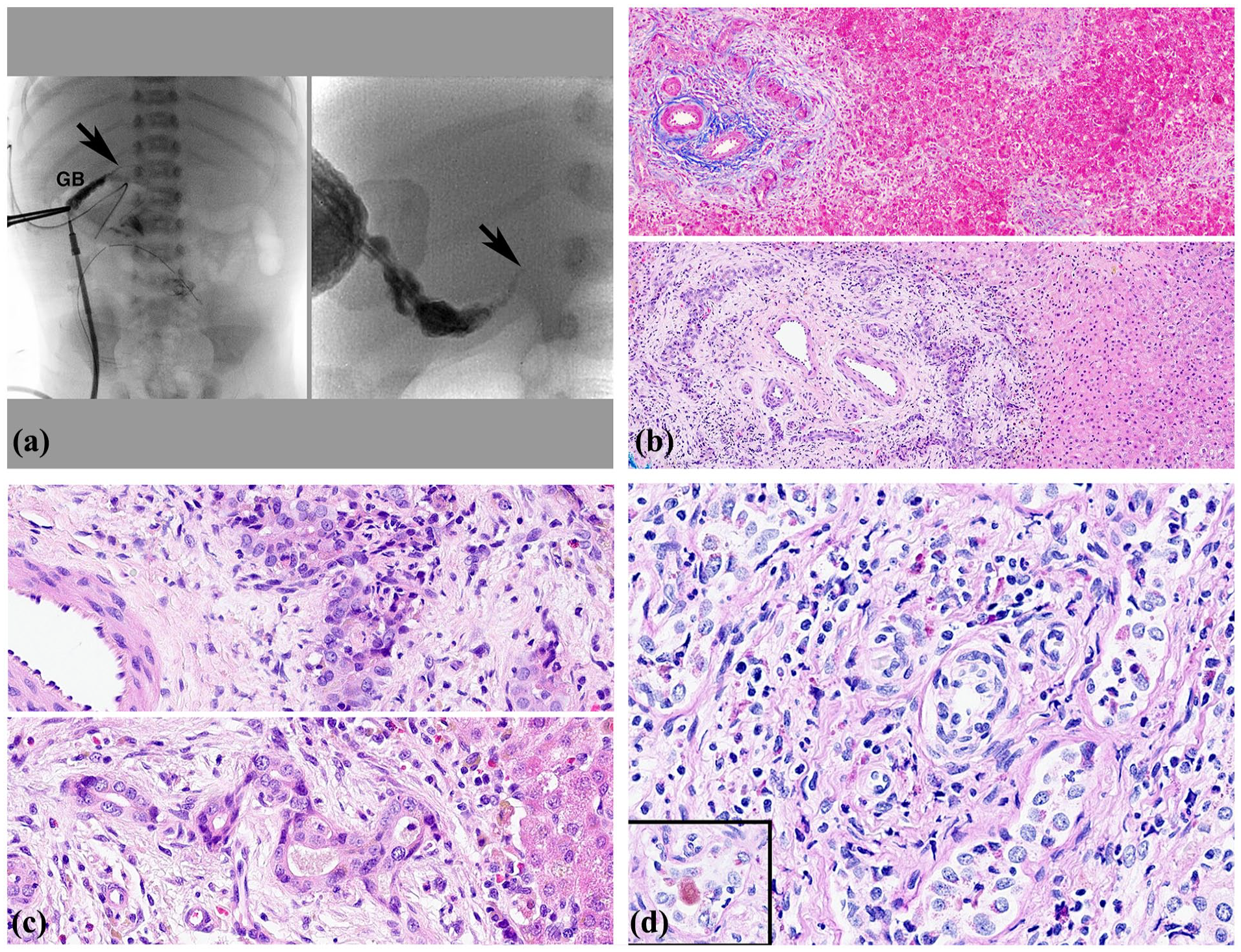
Case#2. (a) Cholangiogram, DOL 30. Left panel: Contrast agent injected at apex of small, contracted gallbladder demonstrates narrow cystic and common bile duct (arrow). There is no visualization of the hepatic duct or intrahepatic ducts. DOL 38. Right panel: contrast injected via the cholecystotomy tube shows narrow cystic and common bile duct (arrow) and a diminutive intrahepatic biliary tree. (b) Upper panel, liver biopsy, DOL 30. Small portal tracts exhibit mild non-specific reactive change. Larger tract contains an incomplete arc of proliferated ducts with local stromal reaction. Masson trichrome ×100. Lower panel, liver biopsy, DOL 39. Most portal areas contain prominent arcs of proliferated ducts in ductal plate configuration with intense cholangitis and loose reactive stroma. H&E ×100. (c) Upper panel. Proliferating ducts with acute cholangitis. Portal stroma is mildly reactive. H&E ×200. Lower panel. Proliferating small ducts show cuboidal cholangiocyte regression with variable reactive nuclei, pale bile, and mild stromal reaction. H&E ×200. (d) Liver DOL 42. Prominent local proliferation of small ducts shows prominent cholangiocyte degeneration with cholangitis. Portal stroma contains scattered ceroid containing macrophages. Inset. A damaged mildly inflamed duct contains PAS reactive pale inspissated bile. PAS-diastase ×200.
On DOL 33, a wedge liver biopsy showed that most small portal areas had mild infiltrates without portal stroma expansion and highly variable reactive changes in small ducts, sparing many but conspicuous in some (Figure 2(b), upper panel). Many larger portal areas had increased duct profiles in a ductal plate configuration with cholangiocyte regression, cholangitis, and reactive portal stroma (Figure 2(b) lower panel and 2(c)). A few portal zones showed prominent increase of bile duct profiles with cholangitis, extreme degeneration of cholangiocytes and increased ceroid-rich portal macrophages (Figure 2(d)).
Weight gain was slow on a regimen of parenteral and oral feeds and enzyme replacement. Jaundice subsided and direct hyperbilirubinemia declined to normal at age 3 months. At age 1 year there is no clinical evidence of liver disease.
Patient 3
A female infant born at term weighed 3.9 kg. The initial newborn evaluation was normal. On DOL 3, abdominal distention due to meconium ileus was relieved at laparotomy with irrigation. Cystic fibrosis was suspected based on sweat chloride of 98 mmol/L and confirmed by demonstration of c.1521_1523del (p. Phe508del) CFTR variant.
TPN was given until DOL 17. Initial mild hyperbilirubinemia resolved with phototherapy by DOL 10 but rose to 4.0 mg/dL on DOL29 when stools were briefly alcoholic. Serum aminotransferase levels were mildly elevated but gamma-glutamyl transferase (GGT) was normal. Ursodiol was briefly administered. The infant gained weight on oral feeds, but jaundice appeared on DOL36. The alpha1-antitrypsin phenotype was MM and the MMP7 serum level was normal. Abdominal ultrasound showed a normal liver, gallbladder, and common bile duct. Percutaneous liver biopsy was performed on DOL 44 to investigate persistent jaundice deemed inappropriate for CF at this age.
The liver biopsy had mild lobular cholestasis with giant cell transformation and mild pericholangitis affecting most small ducts. Active cholangitis and local periportal fibrosis with conspicuous aggregates of pigmented portal macrophages were uncommon; bile duct proliferation was absent (Figure 3). In the next several months, jaundice subsided, hyperbilirubinemia resolved, and a normal pattern of feeding with weight gain was established, at age 3 years, pancreatic insufficiency requires treatment but there is no clinical or laboratory evidence for liver disease.
Case #3. (a) Liver biopsy, DOL 44. Upper panel. Mild pericholangitis consists of mononuclear infiltrate localized to small duct. Lobular cholestasis is mild. H&E ×100. Lower panel. One example of necrotizing cholangitis and uninvolved duct coexist in same portal zone. PAS diastase ×200. (b) Pericholangitis is mild in upper panels (H&E ×400), prominent in left lower panel where ceroid macrophages are markers for leakage (PAS-diastase ×400), and absent in lower right panel (H&E ×400). (c) Ductular reaction without duct proliferation is highlighted. CK7 ×100. Insert: CK7 ×200. (d) Accumulation of ceroid-rich macrophages highlights site of destructive cholangitis. PAS-diastase ×200.
Patient 4
A full-term male infant presented with early onset direct hyperbilirubinemia requiring 5 days in the neonatal intensive care unit (NICU) for monitoring while being breast fed. Direct hyperbilirubinemia persisted at 3.5 mg/dL on DOL 6. Stool color remained normal. On DOL 12, a right upper quadrant ultrasound revealed normal liver without dilatation of ducts. The common bile duct measured 0.1 cm and the gallbladder wall was thick. On DOL 16, hepatobiliary iminodiacetic acid (HIDA) scan was negative for excretion through 24 hours. On DOL 20, an ultrasound-guided percutaneous liver biopsy showed mild lobular cholestasis, portal areas with no inflammation, cholangitis, duct proliferation, or fibrosis (Figure 4(b), upper panel). Focal pericholangitis accompanied by prominent ceroid-laden portal macrophages was uncommon (Figure 4(b), lower panel.)
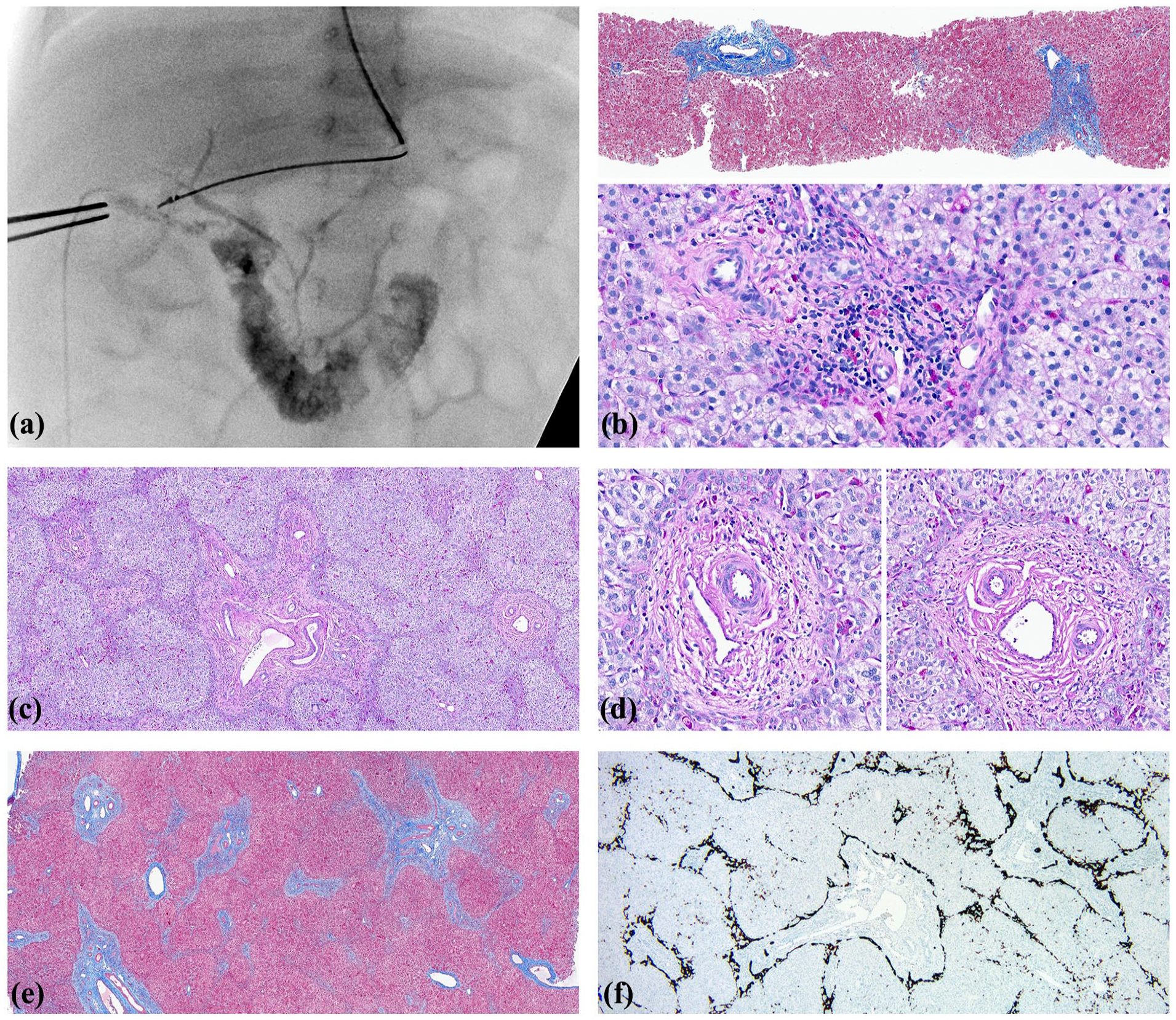
Case #4. (a) Cholangiogram, DOL 50. A small gallbladder, cystic duct, and extrahepatic biliary tree are faintly visualized. Most of the contrast agent has entered the duodenum. (b) Liver biopsy, DOL 20. Upper panel. Lobular architecture is preserved, hepatocyte cholestasis is mild and there is no evidence of cholangitis or bile duct proliferation. Masson trichrome ×50. Lower panel. A site of previous active cholangitis is suggested by chronic pericholangitis and ceroid pigment accumulation in portal macrophages. PAS-diastase ×200. (c) Liver biopsy, DOL 50. Lobular architecture is accentuated by mild portal edema and lobular cholestasis without cholangitis or fibrosis. Most portal zones lack interlobular bile ducts. Portal laminar plate is lined by reactive ductules containing prominent cytoplasmic ceroid pigment. Deposition of ceroid pigment is common in Kupffer cells but absent in portal zones. PAS-diastase ×50. (d) Liver biopsy, DOL 50. Both panels show typical portal areas with a mild ductular reaction and no interlobular bile duct. Ceroid pigment is mostly confined to the ductular reaction and lobular Kupffer cells but not as much as in most portal zones. PAS-diastase ×200. (e) Liver biopsy, DOL 50. Portal areas are slightly expanded but not overtly fibrotic. Masson Trichrome ×50. (f) Liver biopsy, DOL 50. Universal ductular reaction and bile duct paucity is highlighted. CK7 ×50.
He gained weight while breast feeding but direct bilirubin remained elevated on DOL 33. Repeat HIDA scan showed no excretion as before. Direct bilirubin peaked at 4.8 mg/dL on DOL 40. The urine remained clear yellow, and stools were light yellow. Clinical signs of Alagille syndrome were absent. A cholestasis gene panel detected heterozygous CFTR variant: c.454A>T (p. Met152Leu), and homozygous UGT1A1 variant.
At laparotomy on DOL 50, the gallbladder was patent and contained pigmented bile. The liver appeared firm and green. Cholangiogram promptly visualized a small gallbladder, cystic duct, and common hepatic and bile ducts with immediate opacification of the small bowel (Figure 4(a)).
In a wedge liver biopsy lobular cholestasis was uniformly more severe and accompanied by prominent GCT, mild portal zone edema, paucity of interlobular bile ducts, mild ductular reaction at the portal zone margins, and no periportal fibrosis (Figure 4(c)-(f)). Lobular architecture was accentuated by hepatocyte cholestatic swelling and portal zone edema. Frank cholangitis was absent. Moderate deposition of ceroid pigment was common in Kupffer cells but absent in portal zones.
Direct bilirubin levels slowly normalized to 0.16 mg/dL, at 6 months of age. At 9 months of age, he is thriving without medications.
Discussion
We present 4 additional infants with CFTR variants (3 homozygous and 1 heterozygous) and persistent infantile cholestasis; 2 CF homozygotes had intraoperative cholangiograms. Roux-en-Y hepaticojejunostomy was performed for choledochal cyst in case #1. Case #2 had a hypoplastic patent extrahepatic biliary tree; a Kasai procedure was deferred based on findings at laparotomy. In case #4, a heterozygous for CFTR variant and homozygous for Gilbert disease, an intraoperative cholangiogram was normal. Liver biopsies in 3 CF homozygotes showed intrahepatic cholangitis, and portal areas with variable and often severe ductular reaction. A wide range of cholangiocyte injuries including foci of destructive cholangitis. Duct proliferation was highly variable, occasionally simulating that in BA but was without the expansile portal stromal reaction typical for BA. Jaundice slowly subsided in all 4 infants.
We previously observed a similar lesion in liver biopsies from infants with CF including 4 with extrahepatic lesions that mimicked BA. 1 Our overall experience with 10 liver biopsies performed to investigate persistent cholestasis in infants with CF, most with confirmed CFTR pathological variants, is summarized in Table 1, including a heterozygote for CFTR variant coexisting with homozygous mutation for Gilbert disease who did not have active cholangitis (Case #4). ICCF is the basis for persistent cholestasis in the new patients with CF (#1–3 in Table 1). The most active phase of the destructive cholangitis and of the proposed secondary inactive lesion of ICCF are illustrated in images from patients #1, 2, and 3 (Figure 5).
Intrahepatic Cholangiopathy in Infants with Cystic Fibrosis (ICCF).
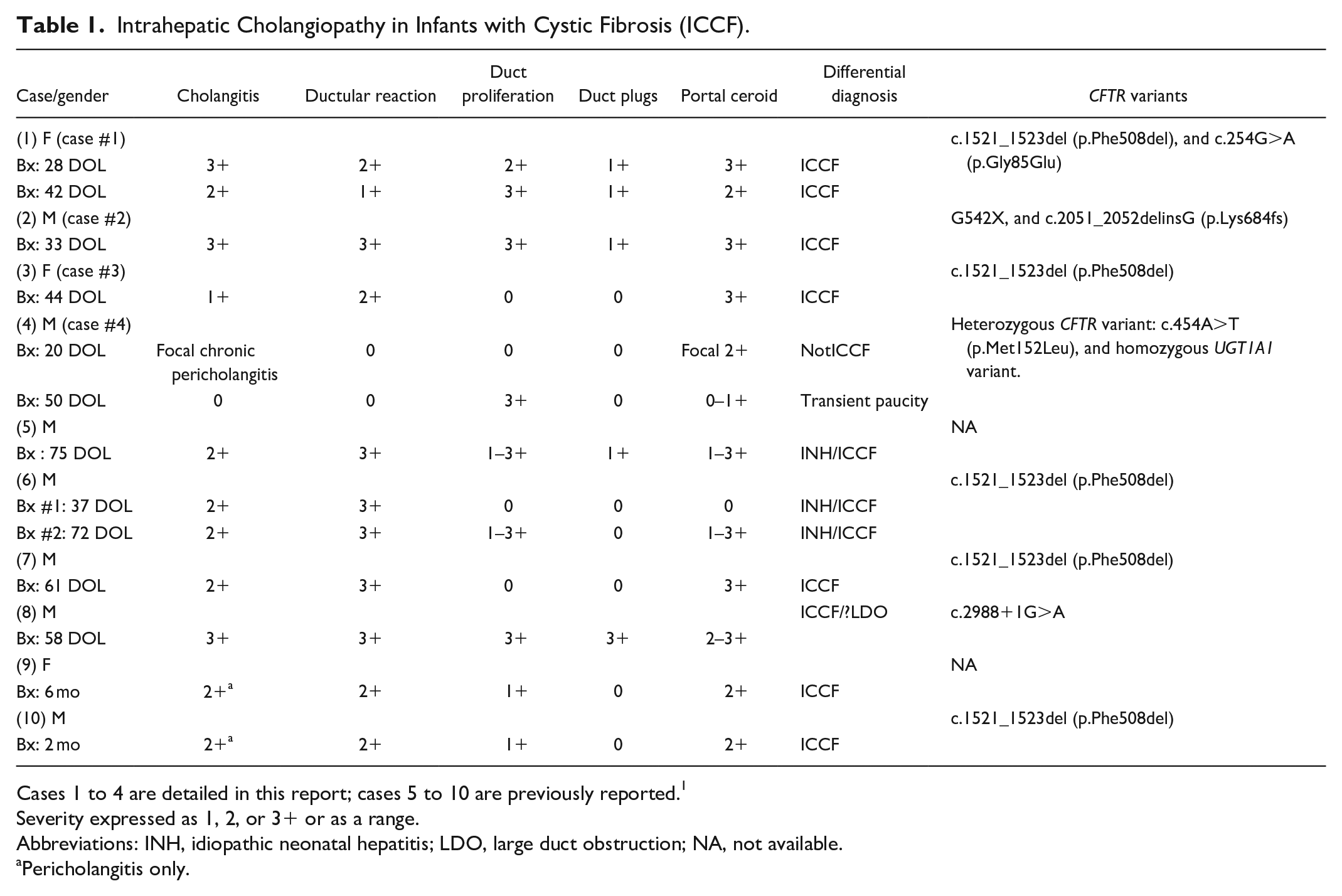
Cases 1 to 4 are detailed in this report; cases 5 to 10 are previously reported. 1
Severity expressed as 1, 2, or 3+ or as a range.
Abbreviations: INH, idiopathic neonatal hepatitis; LDO, large duct obstruction; NA, not available.
Pericholangitis only.
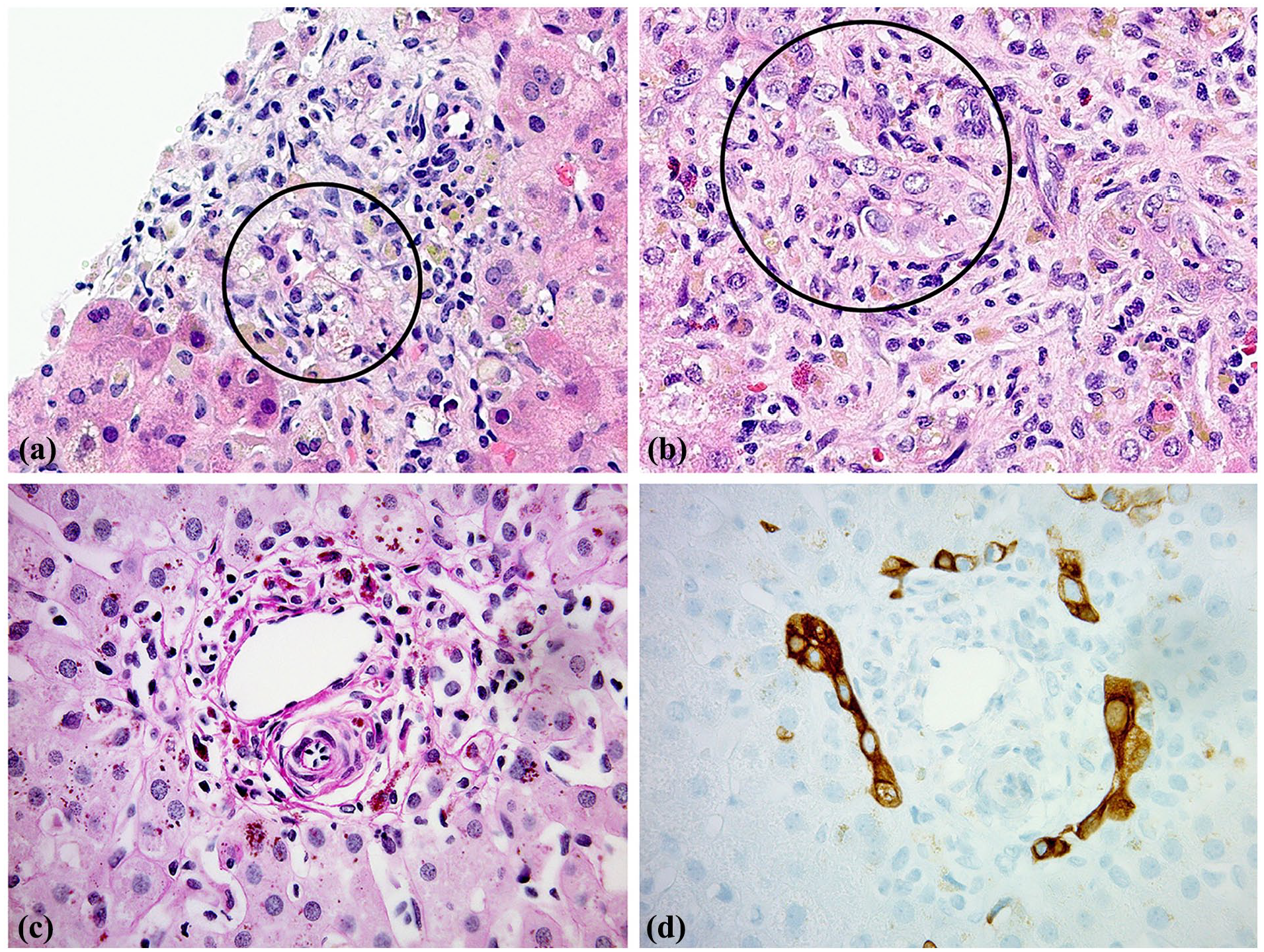
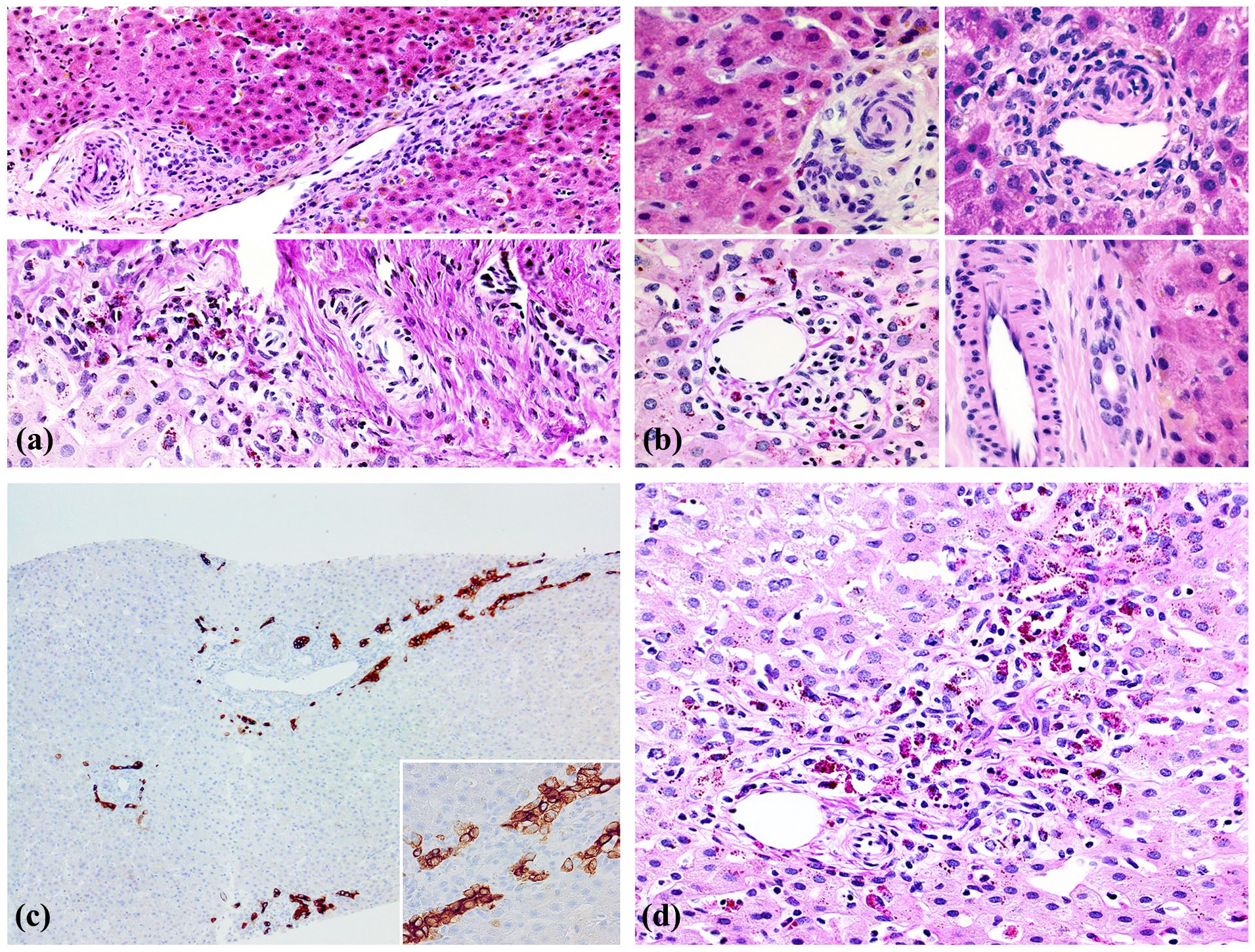
(a) Case #1. Inflamed portal area. Destructive cholangiolitis is highlighted within the circle. H&E ×200. (b) Case #2. Inflamed portal area. Destructive cholangiolitis is highlighted within the circle. H&E ×200. (c) Case #3. Increased portal area cellularity is composed mainly of ceroid-laden macrophages. No bile duct is visible. Pas-diastase ×200. (d) Case #3. Serial section of same portal zone in panel C. Mild ductular reaction is along portal margins. Absence of interlobular duct is confirmed. CK7 ×200.
CF liver alterations are distinguished from those in biliary atresia by the severity of cholangiocyte injury with 1 or more foci of intense cholangitis and cholangiocyte necrosis. Other distinguishing features are absence of expansile reactive portal stroma typical for BA, usually where bile duct proliferation is prominent. Local bile duct leakage producing conspicuous local accumulation of pigmented portal macrophages may occur in syndromic paucity but is non-specific. Unusually pale bile plugs, previously described in intrahepatic bile ducts of infants with CF, though uncommon in these biopsies, are consistent with the idea that the CF bile itself may have unusual qualities. 1
Awareness that a young infant has CF is helped by family history, clinical features, and/or timely genetic information derived from a newborn screen. Investigation of infants with CF and persistent cholestasis may reveal extrahepatic ducts that are either obstructed by mucus or are small and poorly developed. Uniform dilatation of the extrahepatic ducts in patient #1 was classified as a choledochal cyst, not previously reported in CF. Evidence for mucus overproduction was absent in an exhaustive sample of this choledochal cyst wall and gallbladder. Patient #2 had intestinal atresia and meconium peritonitis, common in CF and a recognized risk factor for persistent cholestasis. Transient neonatal hyperbilirubinemia initially subsided in patient #3 but remerged with jaundice several weeks later. Case #4 had normal extrahepatic bile ducts. A large wedge biopsy sample disclosed a universal abnormal pattern interpreted as maturation delay with paucity of intralobular bile ducts but no cholangitis.
Persistent or recurrent cholestasis in infancy typically raises concern at age 2–4 weeks and has multiple causes including BA and many rare genetic diseases. None of our CF patients had clinical signs of bacterial cholangitis or need for prolonged TPN. Since cholestasis in young infants is often transient, liver biopsy is not routinely employed unless hyperbilirubinemia persists and both clinical circumstances and initial laboratory studies do not provide an explanatory context.
In the setting of CF, a putative diagnosis of BA requires a rigorous approach that includes liver biopsy, meticulous intraoperative management and confirmation in tissue excised from the region of the proximal hepatic duct if a Kasai procedure is performed. The possibility of BA is safely minimized once CF has been established by mutation analysis. Serum levels of MMP7 in 2 of our patients did not overlap with the range seen in BA and may prove to be helpful. 5
In infants with ICCF, clinical uncertainty may lead to laparotomy where intraoperative findings appear to be particularly challenging for the surgeon. Surgeons should be aware that poorly developed but patent or unusually narrow extrahepatic bile ducts in CF may result from either low bile flow or mucus obstruction alone or in combination. Intraoperative cholangiography in CF may be aided by flushing of narrow ducts with a mucolytic agent.
Cholestasis in infants with meconium ileus (MI) due to CF is a known risk factor for poor outcome at 1 year of age 6 ; however, there is paucity of liver biopsy-based information about liver disease in infants with CF.1,7 Thus far, jaundice in infants with CF has not been linked to cystic fibrosis liver disease (CFLD) later in life. ICCF appears to slowly regress because direct hyperbilirubinemia spontaneously resolves; long term outcomes are not yet available. Available information about CFLD in older patients with CF includes several longitudinal studies that document clinical characteristics that herald onset without documentation of early histology changes.8-10 The histology of ICCF simulates the accepted pathogenesis for CFLD, that is, recurrent cholangitis, suggesting that CFLD may begin in infancy followed by pruning/remodeling of the intrahepatic biliary tree with progressive focal biliary fibrosis.
We have found no evidence that ICCF is influenced by specific CFTR variants. Extrinsic factors may also influence bile quality and flow in infants such as poor nutrition, dehydration, reduced perfusion, infection, or microbiome imbalance.11,12 Thus, multiple factors may influence the composition of CF bile including lack of normal CFTR function perhaps rendering it unphysiological as it emerges from the lobules to enter the smallest portal ducts. The cholangiocytes of small bile ducts also are selectively injured in defects in bile acid synthesis where toxic intermediary metabolites may cause conspicuous small duct cholangiolitis. 13
Recent introduction of improved genetic animal models for CF that include liver disease, as in the ferret, should lead to improvements in understanding the roles played by each of these factors in pathogenesis of early or recurrent cholestasis and eventually to CFLD and possibly lead to improved therapeutics.12,14-15
In large population studies of patients with CF, specific CFTR variants only partly matched the severity and extent of clinical effects. 16 CFLD typically emerges in older children and adults with CF and has no consistent relationship to the CFTR genotype. One study showed that the risk of severe CFLD reaches 10% at the age of 30 years. 7 Increased risk for CFLD is associated with the SERPINA1-Z allele, history of meconium ileus, pancreatic insufficiency, and CF-related diabetes mellitus. Annual assessment of markers for CFLD in 298 infants identified by newborn screen and followed until age 21 years suggest that risk is associated with AST and GGT values >1.5× the upper limit of normal. 8 A longitudinal study comparing CF patients with and without liver disease from 3 to 21 years old identified noninvasive markers of risk to include platelet count, serum AST and GGT values, and ratios thereof, but relation to noninvasive indices of liver fibrosis was poor. 9
Speculation that heterozygous variants in the CFTR gene may have clinical importance 17 recently acquired a more secure foundation. 18 A comprehensive validated case-control study compared the prevalence of conditions commonly associated with CF in a large population of CF carriers to controls. That study identified 186,000 enrollees that included data from 23,557 patients with CF, 19,802 CF carriers, and 105,000 controls with a 4:1 match of controls to CF or CF carriers. Neonatal jaundice, intestinal atresia, and meconium obstruction were more prevalent in the carrier cohort than in the age-matched cohort of non-CF. Persistent cholestasis in CF infants with heterozygous CFTR variants was not mentioned and has not been reported. Our observation of what appears to be temporary paucity and no ICCF in patient #4, in whom heterozygous CFTR variant coexisted with homozygous mutation for Gilbert disease is of uncertain significance.
It is important to recognize and understand the pathogenesis of ICCF and why it appears to clinically subside with only supportive management. Direct evidence for a cause-and-effect relationship of ICCF to CFLD currently is lacking but the lesion in infants is identical to the active lesion of CFLD in biopsies and explants later in life. Therefore, it is reasonable to propose that ICCF is the prototype lesion for CFLD and that CFLD may begin in early infancy.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.