
Abstract
Urorectal septum malformation sequence (URSMS) is an uncommon disease characterized by a failure of the anorectal septum to divide the cloaca and fuse with the cloacal membrane. Complete URSMS is usually lethal in newborn due to severe renal dysfunction and pulmonary hypoplasia. Partial URSMS is compatible with life with a single perineal opening draining a common cloaca with an imperforate anus which amenable to surgical management. Antenatal diagnosis of URSMS is challenging because of multisystem, complex abnormalities involving gastrointestinal, urogenital tract, cardiovascular, and musculoskeletal systems. In this case report, we describe a 15-week male fetus with partial URSMS having a spectrum of multisystem structural anomalies associated with fetal neuroblastoma in retroperitoneal location and adrenal neuroblastoma in situ.
Introduction
Urorectal septum malformation sequence (URSMS) is a complex congenital anomaly characterized by abnormalities in the migration and fusion of the anorectal septum and the cloacal membrane resulting in anomalies of the urogenital and gastrointestinal tract and occurs 1 in 50,000–2,50,000 newborns.1,2 It was first described by Escobar et al 1 in their series of 6 female infants. 1 Incomplete migration and/or fusion results in persistent cloaca with vesicouterorectal fistula. Persistence of the cloacal membrane results in the absent vaginal and urethral opening and imperforate anus. They are usually associated with ambiguous genitalia because of the failure of normal differentiation of external genitalia.
The entire sequence of malformations is called complete URSMS, characterized by an absent perineal/anal opening, and is usually lethal due to severe renal dysfunction and pulmonary hypoplasia.2,3 Partial URSMS features a single perineal or anal opening draining a common cloaca with an imperforate anus compatible with life.2,3 It is important to differentiate partial from complete URSMS because the prognosis in partial URSMS is generally good, with long-term survival amenable to surgical management.2,3 URSMS can also be associated with renal, cardiovascular, and musculoskeletal abnormalities and brain malformations. This case report describes a 15-week old male fetus diagnosed as partial URSMS with renal and cardiovascular anomalies, pulmonary hypoplasia, and associated retroperitoneal fetal neuroblastoma and neuroblastoma in situ.
Case Report
A 24-year-old primigravida patient with non-consanguineous marriage regularly visited the Obstetrics outpatient department for antenatal check-ups since conception. During her routine investigations and first-trimester scans, there was severe oligohydramnios and nuchal translucency was 1.3 mm at 13 weeks, but the nasal bone could not be visualized at that gestation age due to fetal position. The dual marker test done was in the range of low risk of chromosomal anomalies. After 1 week, the patient was asked to come for repeat ultrasonography in view of severe oligohydramnios and to visualize the nasal bone. However, the patient reported only at 16 weeks for a repeat scan. On ultrasonography, nasal bone was seen, but in addition, multiple fetal anomalies were detected, including a single umbilical artery, non-visualization of bilateral fetal kidney and bladder, severe degrees of oligohydramnios with fetal cardiac anomalies including 2 vessels in 3-vessel view, and over-riding of the aorta in the fetus of 15 + 6 weeks gestation. The case was reported as severe and fatal fetal anomalies. Hence, medical termination of pregnancy was done with 200 mg mifepristone administered on day 1 followed by 400 μg sublingual misoprostol every 4 hours after 48 hours of administration of mifepristone. The patient responded to the treatment and expelled a male fetus with an amniotic sac and placenta.
A complete fetal autopsy was done after obtaining consent from both parents. Fetal weight was 49.05 g, the length 8 cm, and the head circumference 12 cm. The other anthropometric measurements were normal. The umbilical cord showed 2 arteries and a vein. External examination of the fetus shows an imperforate anus. Ears, skull, chest, abdominal wall, and upper limbs were normal. External genitalia is that of the male with a well-formed phallus (Figure 1(a)). Bilateral testicles were seen in the inguinal canal. No skeletal or vertebral anomalies were seen. On cutting open the thorax and abdomen by midline incision extending from the submental region to pubic symphysis, the liver, spleen, pancreas, heart, adrenals, and lungs were visualized in their normal anatomical position. Both the lungs were hypoplastic (right lung: 0.17 g and left lung: 0.06 g). No tracheoesophageal fistula/esophageal atresia was seen.
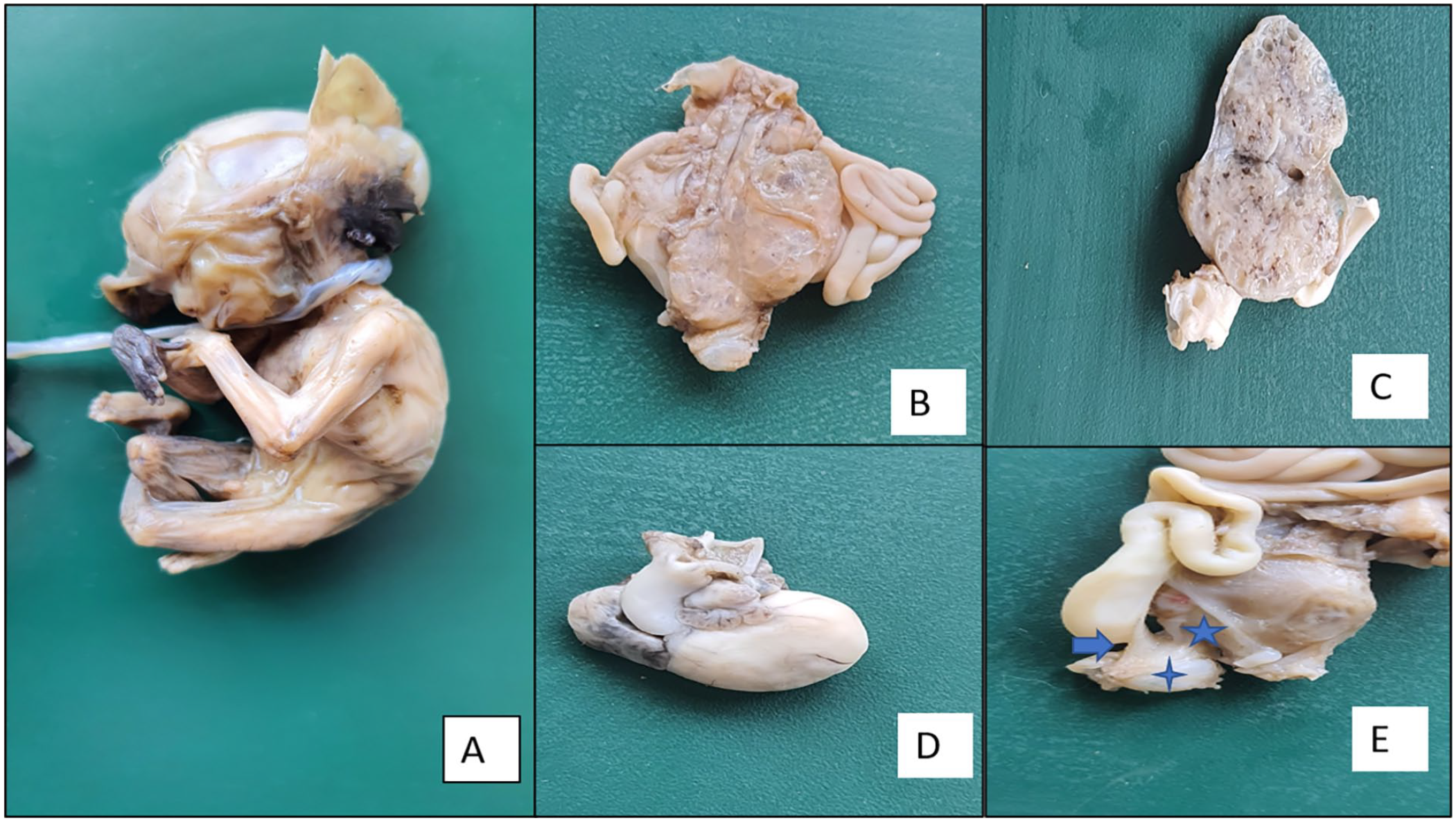
Gross findings. (A) Well-formed male external genitalia. (B) A posterior view of the retroperitoneal organs shows absent left kidney, left ureter, and urinary bladder. The right kidney is enlarged and appears cystic. (C) Cut section of the right kidney—Multiple cysts replacing the parenchyma with intervening fibrous tissue. (D) Enlarged and dilated right ventricle with a single common ventricular outflow tract, the aorta, and main pulmonary artery arising from a common trunk (Type I). (E) Right ureter (star) and large bowel (arrow) draining into the cloaca (diamond).
On dissecting the heart by cutting open the cardiac chambers along the direction of blood flow, there was a single common ventricular outflow tract (Figure 1(d)) with both aorta and main pulmonary artery arising from a common trunk (Collett and Edwards system—type I). The right ventricle appeared enlarged and dilated. Ventricular septal thickness was approximately 2 mm. On dissecting the retroperitoneal organs, the left kidney, left ureter, and urinary bladder was absent. The right kidney was enlarged and appeared cystic (Figure 1(b) and (c)). It measured 1.5 × 0.8 × 0.8 cm3 and weighed 0.68 g with an attached ureter measuring 1cm in length. It drains into the anterior part of the persistent cloaca and drains the sigmoid colon in the posterior aspect (Figure 1(e)). The cut section of the right kidney showed multiple cysts of variable sizes (1–2 mm) with intervening fibrous tissue (Figure 1(c)). Right and left adrenal glands measured 8 × 5 × 5 mm3 (0.09 g) and 8 × 4 × 4 cm3 (0.09 g). No mass lesions were seen in the adrenal glands.
On microscopy, the right kidney showed disorganized renal parenchyma with cysts of variable sizes lined by flattened to the cuboidal epithelium. Islands of undifferentiated mesenchyme were seen with fibromuscular collars (Figure 2(a)) and primitive glomeruli (Figure 2(b)), suggesting multicystic dysplastic kidney disease. In addition, the perirenal tissue showed nodular aggregates of immature neuroepithelial elements with small, blue round cell morphology, Homer-wright pseudorosettes, and evidence of differentiation in the form of ganglion cells and Schwannian stroma (Figure 2(d) and (e)). Overall, features were suggestive of fetal neuroblastoma. Right and left adrenal glands showed fetal adrenal tissue with islands of undifferentiated small blue round cells exhibiting increased nuclear-cytoplasmic ratio and intervening neuropil-like areas, suggestive of neuroblastoma in situ (Figure 2(c)). The conventional karyotyping performed on the fetal cord blood was that of a normal male. However, array-comparative genomic hybridization (array-CGH) was not performed for our patient. The parental conventional karyotyping done was also normal.
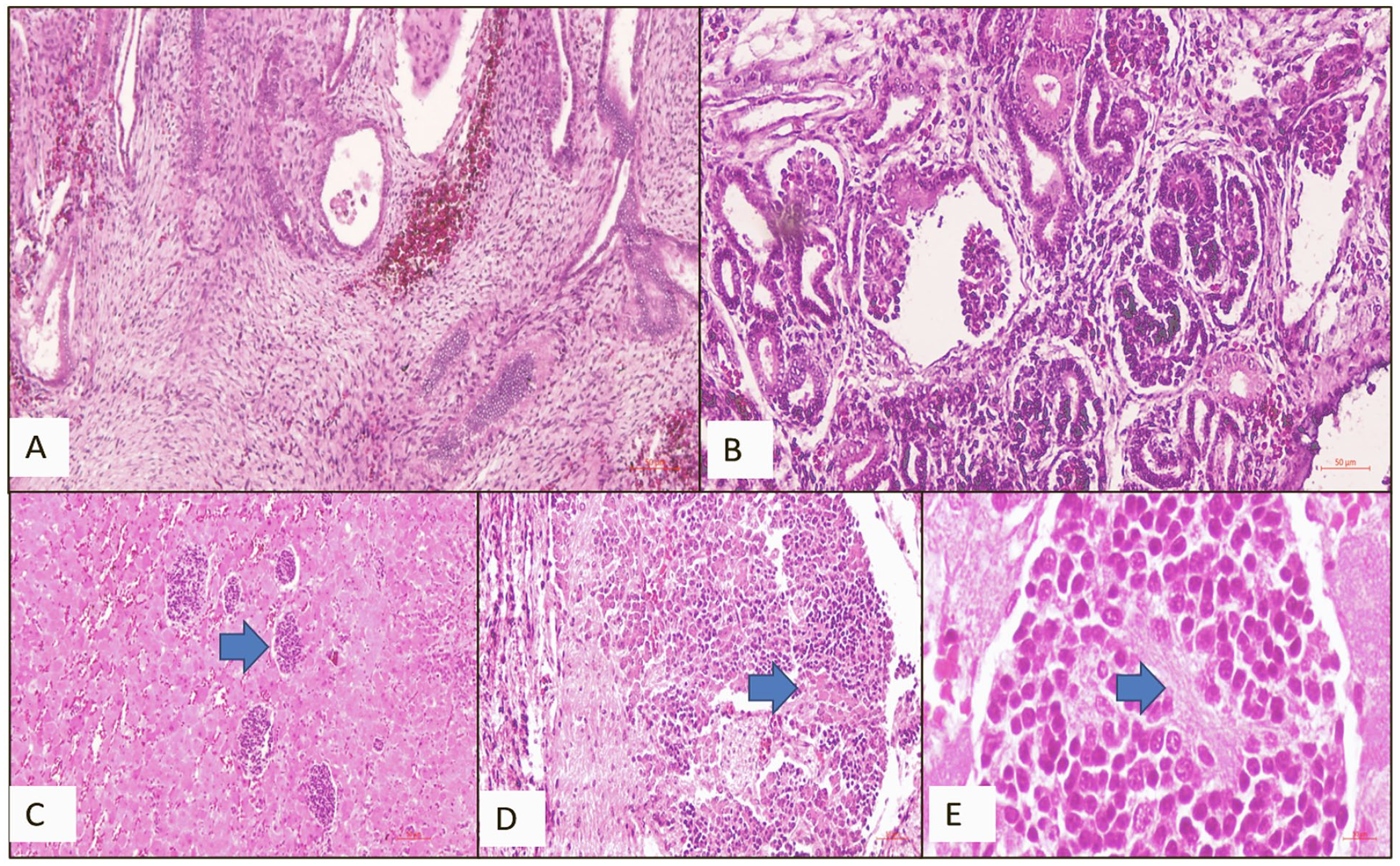
Microscopic findings. (A and B) Multicystic dysplastic kidney disease: Undifferentiated mesenchyme with fibromuscular collars (A) around tubules and primitive glomeruli (B, arrow; H&E ×40). (C) Neuroblastoma in situ: Fetal adrenal tissue with islands of undifferentiated small blue round cells exhibiting increased nuclear-cytoplasmic ratio (H&E, ×40). (D and E) Fetal neuroblastoma: Nodular aggregates of immature small blue round neuroepithelial elements with ganglion cells (D, arrow) and Schwannian stroma (E, arrow) (H&E, D-×40, E-×100).
Discussion
Embryologically, the hindgut initially forms a cloaca bound externally by a cloacal membrane composed of ectoderm and endoderm. The embryology and urorectal septal malformation sequence in females shown in Figure 3. Urorectal septum migrates to the cloacal membrane and divides the cloaca into the ventral urogenital sinus and the dorsal anorectum. 4 The cloacal membrane is divided into the urogenital and anal membranes. The rupture of these membranes results in the patent perineal and anal opening communicating with the amniotic cavity. 2 The caudal mesodermal deficiency was attributed to the incomplete descent of the urorectal septum.5,6 In partial URSMS, incomplete breakdown of the cloacal membrane leads to a single perineal/anal opening and imperforate anus as in our patient. Abnormal external genitalia was suggested to be due to a failure of induction of the early external genital structures and mesodermal tissue formation in the development of these structures. 6 Though etiology is obscure, various genetic events in sonic hedgehog (SHH), homeobox (HOX), PAX, and Ephrin B2 genes have been implicated in interference with the sequential embryological development of the thoracoabdominal and pelvic organs, especially those of the cardiovascular system, urogenital, and gastrointestinal tract.7,8
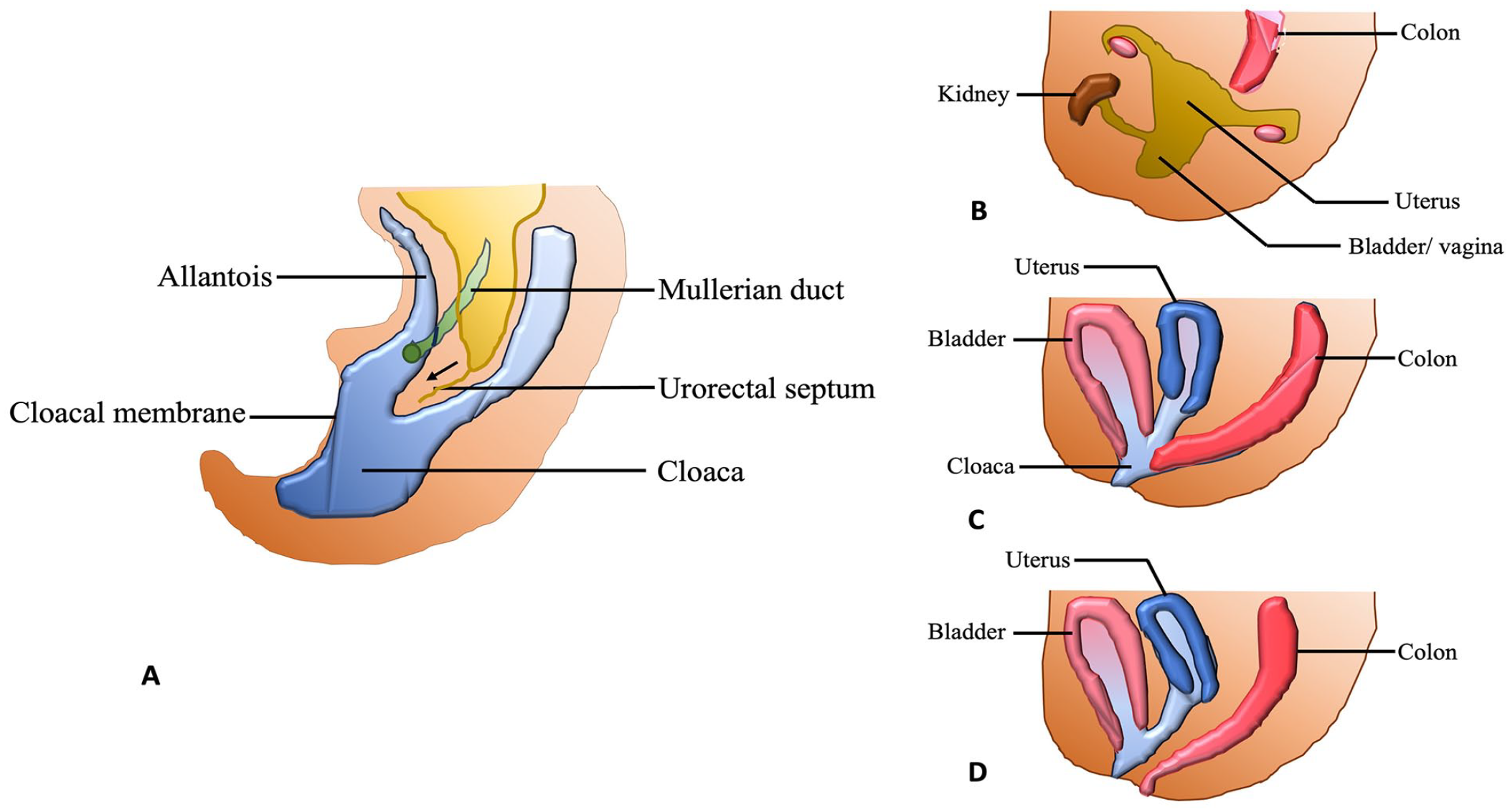
Sketch diagram of embryology and urorectal septal malformation sequence in females. (A) During embryological development, the urorectal septum migrates distally toward the cloacal membrane and divides the cloaca into urogenital sinus in the ventral aspect and anorectum dorsally. (B) Complete URSMS with absent openings of anus and urogenital tract. (C) Partial URSMS with a single opening for anus and urogenital tract. (D) Persistent urogenital sinus with failure of separation of urinary and genital tracts.
Although URSMS was initially reported to be more common in females, it is as common in males. 3 In Wheeler’s partial URSMS series, the male-to-female ratio was 7–18, usually associated with ambiguous genitalia. 2 External genital malformations in less than half of patients with URSMS include hypospadias, a bifid scrotum, penoscrotal transposition, and an absent penis. 2 Our patient had well-formed male external genitalia with normal internal genital organs. Antenatal diagnosis of URSMS is challenging because of multisystem involvement and complex abnormalities involving the genitourinary, cardiovascular, gastrointestinal, and musculoskeletal systems. 9 The presence of urinary tract abnormalities, dilated bowel loops, oligohydramnios, and cystic renal mass are the common antenatal ultrasonographic findings associated with URSMS.4,10 In our patient, non-visualization of bilateral fetal kidney and bladder and severe oligohydramnios were seen. Complete URSMS has a poor prognosis and is incompatible with life. Hence, accurate antenatal diagnosis is vital to allow the termination of pregnancy. URSMS is usually lethal in newborns due to pulmonary hypoplasia resulting from severe oligohydramnios, 3 as observed in the present case.
Other congenital anomalies with URSMS are vertebral anomalies, cranial anomalies, sacral agenesis or hypoplasia, lower limb anomalies, tracheoesophageal fistula, abdominal wall defects, and single umbilical artery.2,3 However, our patient did not have any of these anomalies. In addition, the URSMS needs to be distinguished from other syndromes with similar multisystem involvement, like VATER association and conditions associated with abnormalities in sex hormone synthesis, such as congenital adrenal hyperplasia, especially in those with ambiguous genitalia.4,10 There are similarities in the organ system involvement between VATER and URSMS, but some minor differences exist.10-12 URSMS have a higher prevalence of renal tract defects like renal agenesis, hypoplasia, multicystic dysplastic kidney, anal atresia, and ambiguous genitalia.2,11 Lack of urinary outlet and/or renal agenesis led to oligohydramnios and associated fetal compression resulting in talipes equines varus and pulmonary hypoplasia. Vertebral and limb anomalies in VATER association typically are thoracolumbar hemivertebrae and radial dysplasia, unlike the sacral and lower extremity anomalies in URSMS. 11
Cardiac anomalies in URSMS include atrial and ventricular septal defects and truncus arteriosus.2,4 Our patient had left renal agenesis with an absent ureter and urinary bladder and right multicystic dysplastic kidney disease; anal atresia and truncus arteriosus with both aorta and main pulmonary artery arise from a common trunk (type I). Nervous system abnormalities reported in URSMS include alobar holoprosencephaly, thoracolumbar meningomyelocele, and tethered cord.2,4 None were seen in our index patient. Recurrence of the partial URSM has not been reported in subsequent pregnancies or children of affected individuals. 2 Patients surviving the neonatal period require multiple reconstructive surgeries.
Neuroblastoma is the most common solid tumor in children under 1 year of age. 13 This tumor is derived from neural crest cells and can arise anywhere in the sympathetic nervous system. Antenatal diagnosis of fetal neuroblastoma is made in the third trimester, and adrenals and retroperitoneum locations are the most familiar sites of involvement. 14 Cystic neuroblastomas carry better outcomes compared to non-cystic variants. 14 Widespread use of fetal ultrasonography has led to increased recognition of fetal neuroblastoma. Neuroblastoma in situ represents the collections of neuroblastoma cells in the adrenal gland but rendered distinctive by their microscopic size (>8-µm size), unlike the smaller primitive sympathetic neuroblasts and by the absence of demonstrable metastases.15,16 Usually, it is an incidental finding at autopsy in infants less than 3 months, and it is important to distinguish this from clinically apparent adrenal neuroblastoma. Neuroblastoma has been reported to be associated with persistent cloaca. Cortez et al 17 first reported a 4-year-old patient diagnosed as posterior mediastinal neuroblastoma with persistent cloaca.
In general, it is essential to note an interesting relationship between neuroblastoma and congenital heart defects, seen in up to 20% of the patients.18,19 Major cardiac malformations observed include truncus arteriosus (as in our patient), transposition of the great vessels, and endocardial cushion defects. 14 The treatment options in congenital neuroblastoma include surgery, chemotherapy followed by surgery, or conservative management with ultrasonographic follow-up. Close observation is currently recommended for early-stage neuroblastoma, as spontaneous maturation and remission are well-documented. The use of chemotherapy is for high-stage metastatic disease. 20
Conclusion
As observed in this report of an partial URSMS, the neuroblastoma was likely an incidental postmortem finding associated with multiple congenital anomalies, including cardiac defects like truncus arteriosus. Antenatal diagnosis played a crucial role in managing this condition and aided in differentiating it from similar anomalies involving the urogenital, gastrointestinal, and cardiovascular systems like the VATER association. In our case, a complete autopsy helped confirm the diagnosis and associated malformations.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.