
Abstract
Biliary atresia (BA) is an inflammatory obliterative cholangiopathy which is very common during neonatal and infancy period. We present an autopsy report of a BA in an infant suffering from a genetic syndrome.
Introduction
Biliary atresia (BA) is an inflammatory obliterative cholangiopathy, which is fatal if untreated, usually manifesting in the first months of life. It is characterized by obliteration or discontinuity of the extrahepatic biliary system, resulting in obstruction to bile flow. 1 This disorder can be associated with various congenital abnormalities in 20% of all cases. It represents the most common surgically treatable cause of cholestasis encountered during the infancy. The primary treatments for BA at present are portoenterostomy and, when necessary, liver transplantation.
Portoenterostomy increases the chances of survival, but many long-term survivors may develop complications: in these cases, liver transplantation provides a 90% chance of achieving normal life. 2
We present a case of BA in an infant suffering from a genetic syndrome; the post-operative course was characterized by a fatal complication.
A review of literature was performed both to understand the eventual correlation between BA and the genetic disorder that affected our patient and to investigate the possible complications that determined the failure of Kasai portoenterostomy (KPE).
Case
An 11-month-old female infant has died 9 months after KPE. The girl was born at 38 weeks of gestation from nonconsanguineous parents. Her morphometric parameters were as follows: birth weight 1690 g, height 42 cm, and head circumference 29 cm (<3rd percentile). At birth, micrognathia, right lower limb talus adduct, and thumbs with a fixed palmar flexion were noted. Furthermore, she presented with abnormal facial features, including hypertelorism, long palpebral fissures, long eyelashes with interrupted eyebrows, downturned upper lip, and low-set ears. Doppler echocardiography showed a patent foramen ovale. In addition, funduscopic examination revealed the presence of a “salt-and-pepper” retinopathy.
Chromosome analysis showed a normal 46, XX karyotype. Neonatal period was characterized by marked respiratory distress, feeding difficulties, jaundice, and elevation of transaminase and GGT levels subsequently treated with ursodeoxycholic acid.
At the age of 9 weeks, hepatomegaly, severe and enduring jaundice with acholic stools were noticed, as well as ultrasound findings suggestive for BA. The diagnosis was confirmed with a liver biopsy that showed bridging fibrosis: for this reason, it was decided to perform KPE (time of surgery ~2 hours).
Direct bilirubin levels (DB) before surgery were 13.01 mg/dl; however, after KPE they maintained a steady value (>10 mg/dl). Moreover, the patient suffered from several septic episodes with increase in inflammatory markers attributable to cholangitis.
In subsequent abdominal ultrasound and color Doppler scans, hepatosplenomegaly with signs of portal hypertension was evident. A hyperechoic nodular formation (approximately 10 mm) with hypoechoic margin was present in the region of entero-biliary anastomosis, without dilation of intrahepatic bile duct.
At the age of 8 months, the patient presented a clinical picture of plurimalformative syndrome, facial dysmorphism, microcephaly, spastic tetraparesis, and seizures: therefore, genetic tests were performed and showed a mutation in MLL2 gene and confirmed the suspected diagnosis of Kabuki syndrome (KS).
Afterward, the patient was evaluated for a liver transplant due to worsening of liver function; unfortunately, she developed a multiorgan dysfunction that resulted in a sudden cardiac arrest. Postmortem examination was requested and an autopsy was performed ~24 h after the infant’s death.
At macroscopical examination, the liver was greenish with whitish fibrotic streaks. At the left lobe a nodule (major axis of 1.5 cm) was observed. Furthermore, a polypoid nodule was present on the hepatic side of entero-biliary anastomosis (Figure 1A and B).
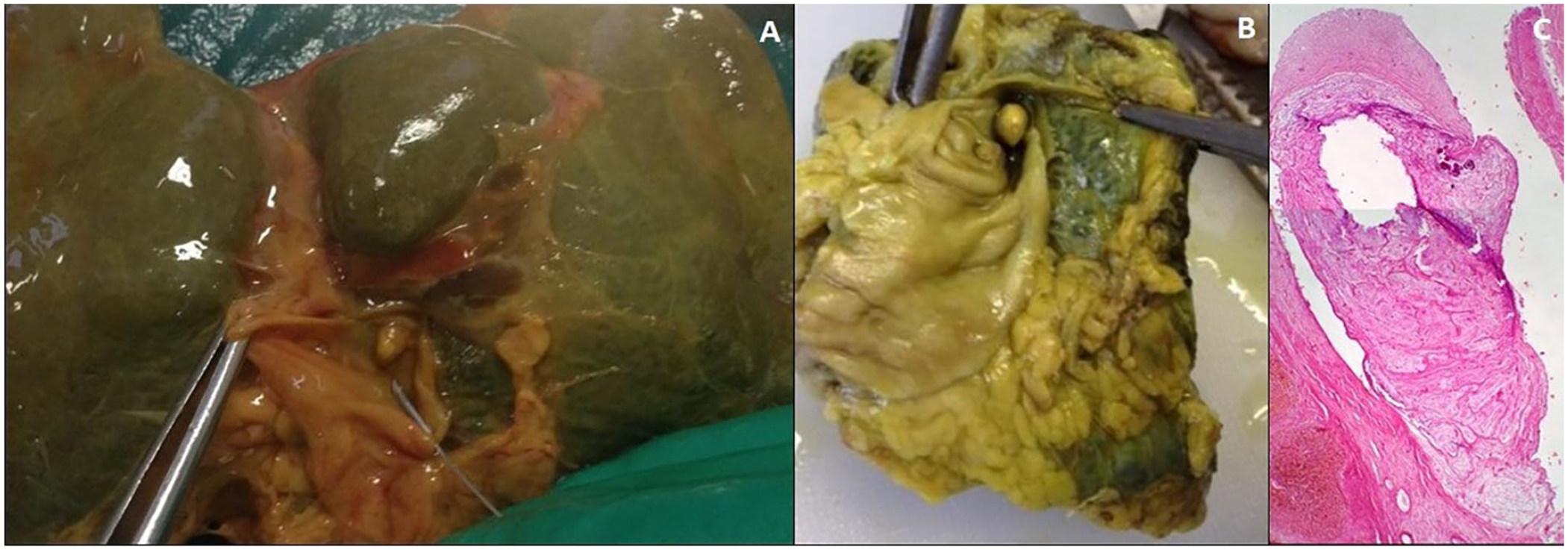
(A and B) Polypoid nodule on the hepatic side of entero-biliary anastomosis observed during autopsy (A) and after fixation (B). (C) Section of liver parenchyma showing a polypoid formation in the middle of biliary-enteric anastomosis with presence of a myxoid stroma.
Histological examination of the polypoid nodule (Figure 1C) showed a myxoid appearance with fibrovascular elements and isolated calcifications of both granular and psammomatous type. Together with the myxoid tissue, loose connective tissue and inflammatory elements were observed.
With regard to the nature of this nodule, the pathologists hypothesized a differential diagnosis between a polypoid myxoma or, much more likely, a polypoid granulation tissue with myxoid degeneration aspects, arisen at the entero-biliary anastomosis. In particular, because of the absence of an epithelial lining, this polypoid nodule was considered a fibro-hyaline mass.
Hepatic parenchyma showed irregular thick fibrous strands around the remaining hepatic tissue, which was characterized by cholestasis signs and biliary thrombi. The fibrous strands revealed angiomatoid-like aspects and a ductular proliferation. For what concerns the 1.5 cm intrahepatic nodule, microscopic examination revealed it was a fibro-hyaline nodule with myxoid elements, compatible with the diagnosis of a mesenchymal hamartoma.
Discussion
Based on clinical picture and genetic tests, we diagnosed KS in our patient. This syndrome is characterized by the combination of craniofacial, dental, and ophthalmologic anomalies, mental and postnatal growth retardation associated with a variety of congenital anomalies (cerebral and musculoskeletal anomalies, disorders of the immune system, etc). 3
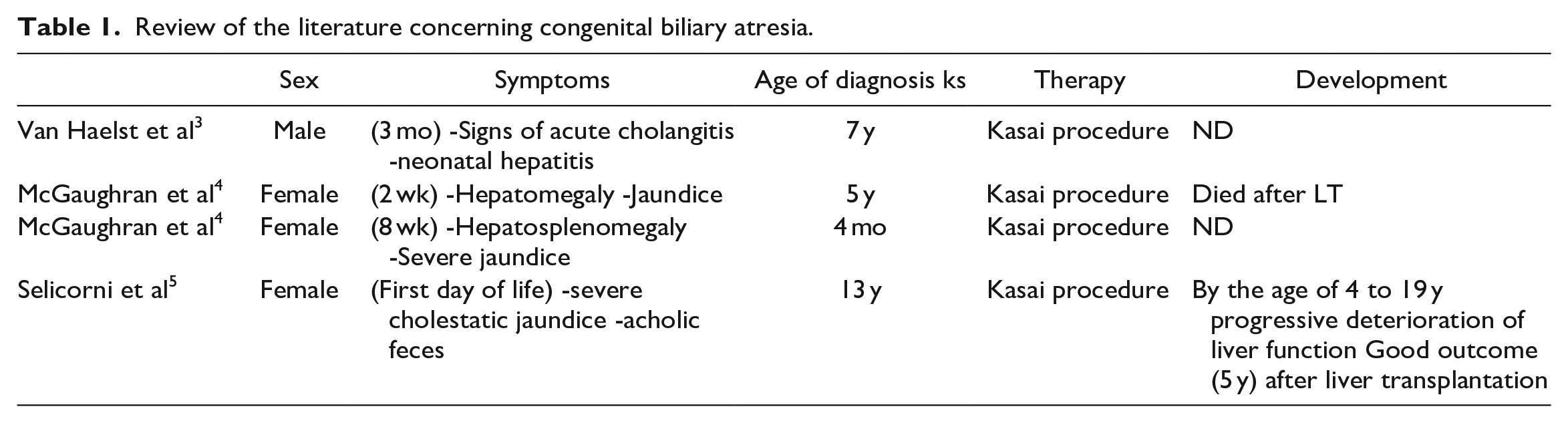
Furthermore, our patient was suffering from BA, which it was promptly treated with KPE. Hepatic anomalies were sporadically described in patients with KS, but the association with extrahepatic BA was observed previously in only 4 cases (Table 1).3-5
Review of the literature concerning congenital biliary atresia.
Our case report confirms that BA can occur in a rare form of KS associated with visceral abnormalities. The presence of cholangitis signs, persistent jaundice or liver dysfunction should arise the suspect of KS, because a diagnosis of BS could be helpful to identify rare forms of KS.
It is important to clarify that bile flow may be inadequate or severe complications can occur after surgery: these are conditions when liver transplantation is urgently needed. Particularly, many studies have shown that recurrent episodes of cholangitis 6 after KPE might suggest an obstruction of the Roux-en-Y loop, determining a poor outcome and shorter survival.
The main causes of obstruction of anastomosis are adhesions, stomal prolapse, and volvulus. 7 Intraluminal obstruction caused by granulation tissue, as in our case, was mentioned only in one dated study, 8 while Chen et al 9 described a case in which the granulation tissue contained pseudohyphae of Candida albicans.
The most important factors in determining a failure of Kasai surgery are bridging liver fibrosis at the time of surgery, 7 and persistence of jaundice after KPE. 1 For this reason, a good pharmacological management should be established for patients suffering from BA. Prophylactic antibiotics can be useful to prevent the onset of recurrent cholangitis. Furthermore, ursodeoxycholic acid can improve bile flow and bile drainage, while steroids (especially in cases that present bridging fibrosis at the time of PE) can reduce and avoid the development of fibrosis or the growth of granulation tissue through their anti-inflammatory and immunomodulatory effects. 2
Conclusion
This autopsy report is unique because of the extremely rare presentation of Kabuki syndrome with biliary atresia. In this particular case, Kasai procedure was unsuccessful due to biliary obstruction by polypoidal fibro hyaline granulation tissue, which had not previously been reported in the literature.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics/Patient Consent Statement
Patient consent was not available; no personal details which may identify the patient, are included in this submission.