
Abstract
The therapeutic window of antibody drug-conjugates (ADC) remains challenging due to safety issues such as interstitial lung disease (ILD) observed with specific deruxtecan-based ADCs. To avoid ILD, we designed M9140 by conjugating the maleimide-containing hydrophilic β-glucuronide linker to exatecan and our anti-CEACAM5 (CarcinoEmbryonic Antigen-related Cell Adhesion Molecule 5) specific antibody. Following repeated iv-infusion at 3 to 30 mg/kg of M9140 every 3 weeks, the pathological findings obtained in cynomolgus monkeys were confined to gastrointestinal and hematolymphoid tissues and resembled the toxicity of exatecan. At 24 mg/kg or higher, transient reductions in neutrophil and reticulocyte counts were observed with each dosing event along with reversible anemia throughout the study. The no observed adverse effect level was 24 mg/kg and the maximum tolerated dose was 30 mg/kg. The difference in toxicity by this small dose increment was correlated with a 2.5-fold difference in plasma exatecan exposure indicating antigen-independent toxicity. As anticipated, no lung toxicity was found with M9140 in these studies that were similar in study design to those used to confirm ILD with trastuzumab-deruxtecan in monkeys. Since the non-human primate model is regarded as predictive for the ILD risk in humans, this result indicates a low risk for ILD when applying M9140 to patients. The current M9140 safety data are discussed with special focus on the absence or presence of ILD with other antibody camptothecin-conjugates, for which a hypothetical pathogenic mechanism is postulated here. The favorable nonclinical profile of M9140 warrants further investigation in patients with CEACAM5-overexpressing tumors.
Introduction
Currently, more than 10 ADCs have been approved for treatment of hematological and solid cancers, yet their therapeutic window remains challenging due to safety issues such as interstitial lung disease (ILD) observed with specific exatecan-derived (DXd)-based ADCs. The mechanism of ADC related ILD has not been elucidated and the topic is specifically addressed here in the context of nonclinical safety assessment of M9140, a novel exatecan-based ADC.
Trastuzumab (T)-deruxtecan (DXd) significantly improved the clinical benefit in patients with HER2-positive metastatic breast cancer (BC) compared to standard of care therapy (e.g., first line treatment with an anti-HER2 antibody and taxane) as well as in patients with this type of tumor being refractory to T-emtansine treatment in second line. 1 Unfortunately, the ILD incidence was 13.6% (25/184 patients) in this clinical phase 2 trial showing durable responses against HER2 (human epidermal growth factor receptor 2) positive metastatic BC at the dose of 5.4 mg/kg T-DXd every 3 weeks (q3w). Recently, patients with HER2-positive metastatic colorectal cancer (mCRC) showed ILD incidences of 8.4% and 12.8% after treatment with 5.4 and 6.4 mg/kg T-DXd, respectively, 2 comparable to 9.6% of ILD at 6.4 mg/kg in gastric cancer (GC) patients. 3 The overall incidence of adjudicated drug-related ILD/pneumonitis was 15.4% in a pooled analysis of nine monotherapy studies of T-DXd with 1150 patients in total. 4 This analysis included 2.2% of fatal cases and resulted in a prescription box warning for ILD requiring careful monitoring of respiratory symptoms and discontinuation of treatment in all patients with grade 2 or higher ILD/pneumonitis. 5 This calls for a better understanding of the possible cause of ILD when designing ADCs with DXd or related payloads.
From 2019, topoisomerase 1 inhibitor-based ADCs such as T-DXd and anti-TROP2-SN38 were successfully introduced for targeted treatment of cancer. Topoisomerase 1 inhibitors belonging to the class of camptothecins such as SN-38 (7-ethyl-10-hydroxycamptothecin, the active metabolite of irinotecan), exatecan, and DXd, a derivative of exatecan (2-hydroxyacetyl exatecan), are known to mainly induce myelosuppression (specifically neutropenia) and gastrointestinal (GI)-tract toxicities by their indirect DNA-damaging and apoptotic activity, 6 but do not cause lung toxicity.7-10 DXd alone did not induce lung toxicity in monkeys and rats when dosed repeatedly (5x weekly), despite much higher DXd plasma exposure than that of liberated DXd in the monkeys given T-DXd. 11 This indicates the ILD risk of DXd-based ADCs cannot be explained by the toxicity of the payload directly.
The monkey model is regarded as predictive for the risk of ILD in human. 11 In the monkey, ILD is manifested by inflammatory cell infiltrates (such as neutrophils and lymphocytes) in the alveolar wall, intra-alveolar fibrosis, alveolar edema, and aggregates of foamy alveolar macrophages. 11 Apparently, the pathological process of ILD needs time to develop given the increased incidence of ILD (from about 10% to 40%) and reduced level of the highest non-severely toxic dose (HNSTD) (from 78.8 mg/kg to 30 mg/kg) when dosing monkeys either for 1.5 (3x q3w) or 3 month (5x q3w) with T-DXd. Interestingly, pulmonary toxicity has not been observed in monkeys when targeting HER2 with the same Ab (T) but bearing a different linker-payload (L-DM1), called T-emtansine (3x q3w). 12 In studies with T-DM1 (q3w) of 3 months, only mononuclear cell infiltration of the interstitial lung was found in monkeys which was consistent with low levels of pneumonitis (below 1%) in patients treated with T-emtansine. 11 In addition, HER2 expression in monkey and human lungs is restricted to the bronchial level, while T-DXd lesions were found at the alveolar level of the lower lung lobes in monkeys. Together, these data do not point to a specific role of HER2 in the manifestation of ILD.
Notably, ILD has also been observed with identical DXd-based ADCs such as the trophoblast cell surface antigen 2 (TROP2) (Datopotamab-DXd)13,14 and HER3 (Patritumab-DXd). 15 In contrast, ADCs containing SN-38 targeting TROP2 or CEACAM5, respectively, sacituzumab govitecan16,17 or labetuzumab govitecan, 18 do not induce pulmonary toxicity. No lung toxicity/ILD was noted in 86 patients with r/r mCRC dosed with labetuzumab govitecan (CEACAM5-SN-38). 18 Also, TROP2-SN38 (sacituzumab govitecan) did not cause lung toxicity in monkeys 16 and only one case of grade 3 pneumonitis was found—but no ILD—in the TROP2-SN38-dosed group of 258 patients with metastatic triple negative BC. 17 Despite the relatively labile ADC in plasma due to themaleimide-CL2A linker-SN38 technology, these results suggest that CEACAM5, TROP2, or the payload SN38 do not seem to play a specific role in the etiology of ILD. Meanwhile, other ADCs have been designed targeting HER2 and TROP2 to liberate exatecan-derivatives with enzymatically cleavable maleimide-tetrapeptide linkers that were not associated with ILD in repeat-dose monkey toxicity studies.19–21 Together these data suggest ILD is both target- and payload-independent and allude to an apparent unique property of DXd-based ADCs from the same technology platform known to induce ILD.4,13,15 It is hypothesized in this paper that methylamine, the small spacer in the specific linker-payload construct that is co-released with DXd from the ADC, 22 may mediate ILD.
To avoid ILD, M9140 was manufactured by conjugating the maleimide-containing hydrophilic β-glucuronide linker directly to exatecan and our anti-CEACAM5 (CarcinoEmbryonic Antigen-related Cell Adhesion Molecule 5) specific antibody with a drug-to-antibody (DAR) ratio of 8 (Figure 1). Exatecan alone has potent antineoplastic activity in a broad range of tumors but showed dose-limiting hematotoxicity (e.g., myelosuppression), and intestinal toxicity in clinical studies.7,8,23,24 The current M9140 ADC concept aims to improve the clinical therapeutic window by targeted release of the cytotoxic payload exatecan into the CEACAM5 overexpressing tumor(s) without introducing ILD. In nude mice (NU/NU) bearing patient-derived colorectal cancer xenografts, a single treatment of 10 mg/kg M9140 induced tumor growth inhibition in 10 models and tumor regression in 4 models displaying 66%–95% tumor volume inhibition as best response.
25
Here we investigated and report the safety profile of M9140 obtained from repeat-dose toxicity studies in cynomolgus monkeys that were similar in study design to those used to confirm ILD with T-DXd.
11
Currently M9140 is dosed to patients with advanced stage or metastatic CRC for evaluation of safety, tolerability, and pharmacokinetics (ClinicalTrials.gov Identifier: NCT05464030). The chemical structure of M9140. M9140: Exatecan linked to an enzymatically cleavable, hydrophilic β-glucuronide moiety, and maleimide conjugation with a DAR of 8 to anti-CEACAM5 IgG1.4 mAb.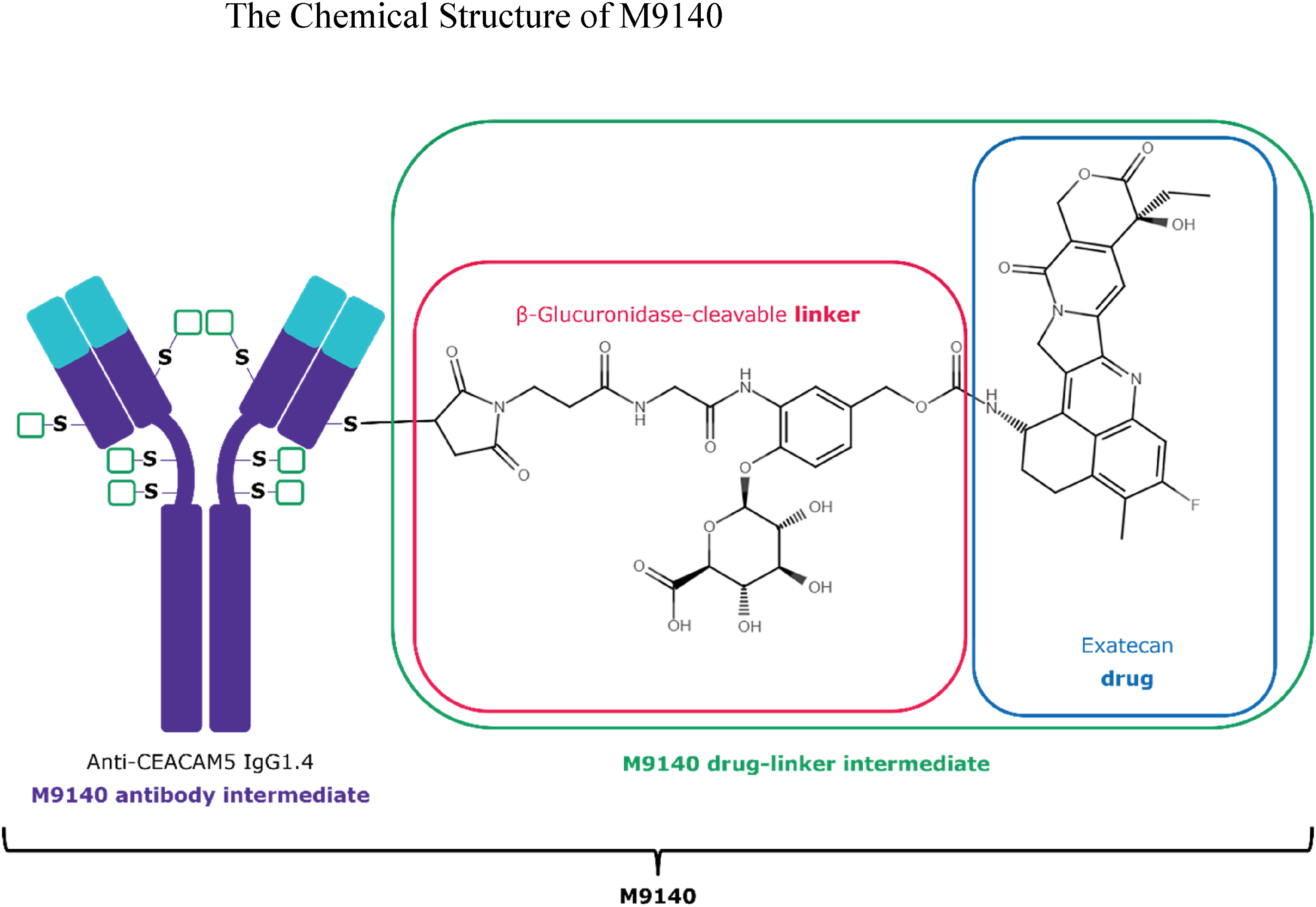
Methods
All procedures in animals described below were performed in compliance with the Animal Welfare Act(s), the recommendations of the AAALAC and national Animal Health regulations. Protocols were reviewed by the Institutional Animal Care Use Committee of Merck KgaA (Darmstadt, Germany), and Istituto di Ricerche Biomediche “Antoine Marxer”–RBM S.p.A. (Ivrea, Italy). The monkeys were allocated into experimental groups based on their individual behavior and history. Room temperature and relative humidity were monitored continuously. For scheduled or unscheduled necropsies, monkeys were terminally sedated with an overdose of intravenous injection of barbiturate (50 mg/kg) after having received a parenteral injection of a muscle relaxant and analgesic agent (xylazine—Rompum® 0.05 mL/kg).
Purpose-bred, naïve cynomolgus monkeys (Macaca fascicularis) were purchased from Envigo (Horst, The Netherlands). Monkeys were born and bred in Vietnam and quarantined in Camarles (Camarney SLU, Spain). At the test facility (Istituto di Ricerche Biomediche “Antoine Marxer”—RBM, Ivrea, Italy), monkeys were housed in an air-conditioned room (22 ± 2°C), with 15–20 air changes per hour, a relative humidity of 55 ± 15% and artificial lighting with a circadian cycle of 12 hours from 7
Intravenous Dose Formulations
M9140 (Ab manufactured by EMD Serono, Billerica, USA or by Merck Aubonne, Switzerland, and conjugated by MilliporeSigma, St. Louis, MO, USA) was formulated in a vehicle solution (control groups) containing 10 mM histidine, 233.7 mM trehalose dihydrate, 5 mM L-methionine, 0.05 % polysorbate 20 (pH 5.5) and stored protected from light and refrigerated (2–8°C) until use. The stock concentration (10 mg/mL or 15 mg/mL) and dilutions of M9140 were verified for stability and concentration prior to dosing (Chemical & Pharmaceutical Development/Analytical Development, Merck KGaA, Darmstadt, Germany). The test item or vehicle was administered by 30-minute intravenous (iv) infusion to cynomolgus monkeys (saphenous vein). Dose formulations of all dilutions kept from the pivotal toxicity study were verified by a validated analytical method (HPLC) and remained within the acceptance criteria (±15%, RBM, Ivrea, Italy).
General Parameters for the Evaluation of Toxicity
Toxicity indices consisted of clinical observations, body weight, food consumption, ophthalmology, clinical pathology (clinical chemistry, hematology, coagulation, urinalysis), immunophenotyping, gross pathology, organ weights, and histopathology. Necropsies included examination of the carcass; external body orifices; abdominal, thoracic, and cranial cavities; and organs. Tissues collected at necropsy were preserved in 10% neutral-buffered formalin, Davidson’s (eye and optic nerve) or modified Davidson’s (testis) fixative and were processed for routine histologic examination (paraffin wax-embedded tissues and HE stain). Selected organs were weighed prior to fixation. Additionally, unscheduled necropsy and histology was performed on moribund animals. Necropsy for macro- and microscopic examinations was scheduled on day 50, 1 week after the final dose, and on day 71 for recovery animals.
Repeat-Dose Monkey Toxicity Studies with Concomitant Toxicokinetic Evaluation
In the pilot study, M9140 was administered every 3 weeks (q3w) by 30-minute (±5 minutes) i.v. infusion to 1 male and 1 female monkey per group at 0, 3, 10 and 30 mg/kg for 3 consecutive times (day 1, 22 and 43). One additional male monkey was infused at 60 mg/kg to explore the maximum tolerability. In the pivotal study, M9140 was similarly infused to monkeys at 0, 6, 12, and 24 mg/kg (q3w x3) to 3 subjects/sex/group and additionally 2 animals/sex in the control and high dose group for recovery evaluation.
Mortality and clinical signs (physical appearance, behavior, and general signs) were recorded twice daily and at the end of the infusion (until 30-min post-dose), and body weight was recorded once weekly from 1 week before commencement of treatment throughout the study period. Food and water intake was conducted daily (per cage). Ophthalmoscopic investigation was done once pre-treatment, at the end of the last infusion, and during the recovery period. Hematology (ADVIA2120i Siemens analyzer) and clinical chemistry (AU480 Beckman Coulter analyzer) were investigated in all monkeys during the pre-treatment period, and on days 3, 8, 22 (before second dosing), 24, 29, 43 (before third dosing), 45 and 50 (before sacrifice), and during the recovery phase (Day 57 and 71). Immunophenotyping (FACLyric Becton Dickinson flow cytometer, Becton Dickinson antibodies) was performed on the same days to measure absolute and relative counts of total B cells (CD3−, CD20+), total (CD3+), helper (CD3+, CD4+) and cytotoxic T cells (CD3+, CD8+) and natural killer cells (CD3−, CD16+). For all monkeys, coagulation parameters (prothrombin time and activated partial thromboplastin time) and standard urinalysis were evaluated pre-treatment, on day 50 (before sacrifice), and at the end of the recovery period (day 71).
Blood samples (0.6 mL in lithium heparin tubes) for bioanalysis and toxicokinetic (TK) evaluation were collected from each animal on Days 1 and 22 (pilot study only) and at the following time-points: 0 (pre-dose), and 0.5 (= end of infusion), 2, 6, 24, 48, 96, 168, 240, 336, and 504 h post-infusion (before next dose), and for Day 43 on the same timepoints up to 168 h in each animal, and in addition on 240, 336, and 504 h in recovery animals only.
Integrated Safety Pharmacology Investigations
As an integral part of the pilot and pivotal monkey toxicity studies, heart rate (HR), electrocardiogram (ECG), and arterial blood pressure (systolic and diastolic BP), were measured in all monkeys at baseline (pre-dosing) and at 0.5 h (±20 min) and at 4 h (±20 min) post-infusion on Day 43, and once during the last week of recovery (Day 67 or 70). Animals were trained to undertake recordings under conscious, temporarily restrained condition while sitting in a chair. Three consecutive measurements of systolic and diastolic arterial blood pressures were collected before respiratory rate and the ECG measurement session. Evaluations were performed on at least 10 representative ECG complexes. In the pilot study, respiratory rate was measured immediately after arterial blood pressure measurements by counting the respiratory movements over 1 minute. A clinical neurological assessment was made in conscious animals (all subjects) in their home cages during the pivotal study at the following time points: once during the pre-treatment period (Day 1 or 5), at 3 h post-infusion (±30 min) on Day 43, and once during the last week of recovery (Day 67, 68, or 70). The following neurological parameters were evaluated in all animals: number of perching (number observed during the observation period), lethargy/arousal, posture, salivation, tremor activity, convulsive activity, movement of facial muscles, eyelid closure, lacrimation, piloerection, gait, gait score, bizarre behavior, spontaneous vocalization (presence or absence), pupil response (presence or absence), and approach response. The parameters were recorded according to a scoring system or described as indicated above in parentheses.
Rectal temperature was recorded in all animals of the pilot study: twice pre-dosing, and on days 1, 22, and 43, each at 0.5 h (within 30 minutes post-dose) and at 24 hours (±30 minutes) post-dose. In the pivotal study, all animals were recorded once during the pre-treatment period (Day 1), at 0.5 h (±20 min) and at 4 h (±20 min) post-infusion on Days 1, 22, and 43, and once during the last week of recovery (Day 67, 68, or 70).
ECG electrodes (single use foam electrodes) were placed on each animal according to Standard II Leads. The signals were digitized (A/D converter ACQ-7700, DSI, St. Paul, Minnesota, USA) and continuously recorded once the heart rate reached a steady state, using a software package (Ponemah Physiology Platform 5.20, from DSI supplier). A cuff for arterial blood pressure (BP) measurement was connected to a blood pressure monitor (Monitoring vital signs unit CARESCAPETM V100, GE Healthcare, Milwaukee, Wisconsin, USA) and placed on one of the forearms.
No statistical analyses were performed in the pilot study due to the low number of animals per group. Where appropriate in the pivotal study, absolute body weight, clinical pathology parameters, ECG data, blood pressure, body temperature, respiratory rate, and organ weights—relative and absolute—of each dose group were compared with those of the control group, using the multiple 3-sided Dunnett test. Taking the number of dose groups into account, all test procedures used maintain a multiple significance level of P ≤ 0.05.
Methods of Analysis for Toxicokinetic Evaluation
To support TK assessment of the pivotal and pilot toxicity studies in monkeys, validated or qualified bioanalytical methods were used for the quantitative determination of M9140 analytes (total antibody, conjugated antibody, and unconjugated exatecan) in plasma. For quantification of total antibody (conjugated and unconjugated), a biotinylated recombinant human CEACAM5 protein was used as a capture reagent and a ruthenylated (SULFO-TAG-conjugated) anti-human IgG-heavy and light chain monkey-adsorbed antibody was used as a detection reagent. The lower limit of quantification (LLOQ) of the assay was 100 ng/mL. For the quantification of conjugated antibody (or ADC), a biotinylated anti-exatecan antibody was used as a capture reagent and an Alexa Fluor-647 labeled anti-idiotype against anti-CEACAM5 antibody was used as a detection reagent. The assay LLOQ was 100 ng/mL. A validated ultra-performance liquid chromatography with tandem mass spectrometry (UPLC-MS/MS) method was used to measure unconjugated exatecan. Plasma samples were extracted using protein precipitation technique followed by chromatography using a Waters ACQUITY™ I-Class plus UPLC™ system equipped with an ACQUITY™ UPLC™ BEH® C18 2.1 × 100 mm, 1.7 μm, column. A Sciex triple quad™ 5500 mass spectrometry equipped with turbo ion spray source operating in positive ion mode was used for detection. Samples were quantified by using isotopically labeled exatecan as internal standard. The method concentration range was from 0.100 ng/mL to 10.0 ng/mL.
Toxicokinetic parameters were determined from plasma concentrations versus time data obtained from each monkey using non-compartmental analysis in Phoenix WinNonlin® version 6.3 (Pharsight Corporation, USA). TK analyses were conducted using the linear log trapezoidal calculation method. The parameters of interest were the area under the concentration-time curve from 0 to 168 h (AUC0–168 h) and from 0 to 504 h (AUC0–504 h), maximum concentration (Cmax), time to reach maximum concentration (tmax), and half-life (t1/2). AUC0–168 h and AUC0–504 h were used to assess dose proportionality of each analyte.
Assay to Detect Anti-Drug Antibodies
A qualified immunoassay method was used for the detection of antibodies against M9140 in cynomolgus monkey serum. Briefly, an acid treatment dissociated the complexes in the study sample and subsequent neutralization allowed the detection of anti-drug antibodies in the presence of free drug. M9140 conjugated with biotin was used as the capture reagent, and M9140 conjugated with SULFO-TAG was used as the detection reagent. The drug tolerance level was 20 and 1000 μg/mL at the low positive control and high positive control concentrations, respectively. The rabbit antiserum from animals immunized with the ADC to obtain anti-ADC C2546 IgG (batch lot 81004 produced at BioGenes GmbH, Berlin, Germany) was used as positive control.
Results
Toxicity Studies with Concomitant Toxicokinetic Analysis in Monkeys
Pilot 7-Week Toxicity Study
Iv-infusion with M9140 up to 30 mg/kg once every 3 weeks showed only occasional slight diarrhea and mild body weight fluctuations at 30 mg/kg, except for the female after the last administration of 30 mg/kg. After the third time of dosing, this female showed generally moderate clinical signs (hypoactivity, diarrhea, hypothermia, diffuse piloerection) at day 49–50 and body weight loss (−14.5%) at day 50 (scheduled sacrifice). Dosing at 60 mg/kg resulted in premature sacrifice of the male monkey on day 9 due to ethical reasons concerning adverse clinical signs (as above) and body weight decrease of −10.5% on the day of premature sacrifice. In addition, clinical pathology examinations of the preterm animal on day 8 revealed decreased pancytopenia, albumin, sodium, and calcium, and increased liver enzymes (alanine and aspartate transaminases, glutamate dehydrogenase), creatinine, and c-reactive protein. Elevations of plasma liver enzymes were in absence of any histological hepatic changes.
At 30 mg/kg/occasion, a transient decrease of neutrophils in males and reticulocytes in both animals were observed after most treatments (up to about −80% to −90% at nadir on Day 7; Figures 2A and B). A decrease in red blood cell mass (about −20%) was also evident in the female after the third treatment (Figure 2C). At 10 mg/kg/occasion, decreased neutrophils (up to −66% after the third infusion only) and reticulocytes (up to −83% after each treatment) were observed only in the male animal. No relevant changes were seen at 3 mg/kg/occasion. (A) Time-course of neutrophil counts following repeated iv dosing of M9140 (q3wk) to cynomolgus monkeys. (B) Time-course of reticulocyte counts following repeated iv dosing of M9140 (q3wk) to cynomolgus monkeys. (C) Time-course of erythrocyte counts following repeated iv dosing of M9140 (q3wk) to cynomolgus monkeys. Time-course of mean neutrophil (A), reticulocyte (B), erythrocyte (C) counts/dose group and depicted per gender (plots a and b) following 0, 6, 12, 24, and 30 mg/kg M9140 every 3 weeks for 3 times (d1, d22, d43) in male (M) and female (F) monkeys. The group size was 1/sex at 30 mg/kg, 3/sex at 6 mg/kg, and 5/sex at 0, 12, and 24 mg/kg. Based on internal historical control data from 73 male and 72 female cynomolgus monkeys of Vietnamese origin mean±SD (2.5%−97.5% percentiles) of control laboratory reference ranges were as follows: erythrocytes in M: 6.15 ± 0.43 106/μL (5.39–6.94) and F: 5.93 ± 0.45 106/μL (4.97–6.77); reticulocytes in M: 75.5 ± 19.7 109/L (36.5–117.1) and F: 73.8 ± 25.0 109/L (31.9–131.6); neutrophils in M:5.91 ± 3.05 103/μL (1.94–12.60) and F: 6.65 ± 3.03 103/μL (2.60–13.32).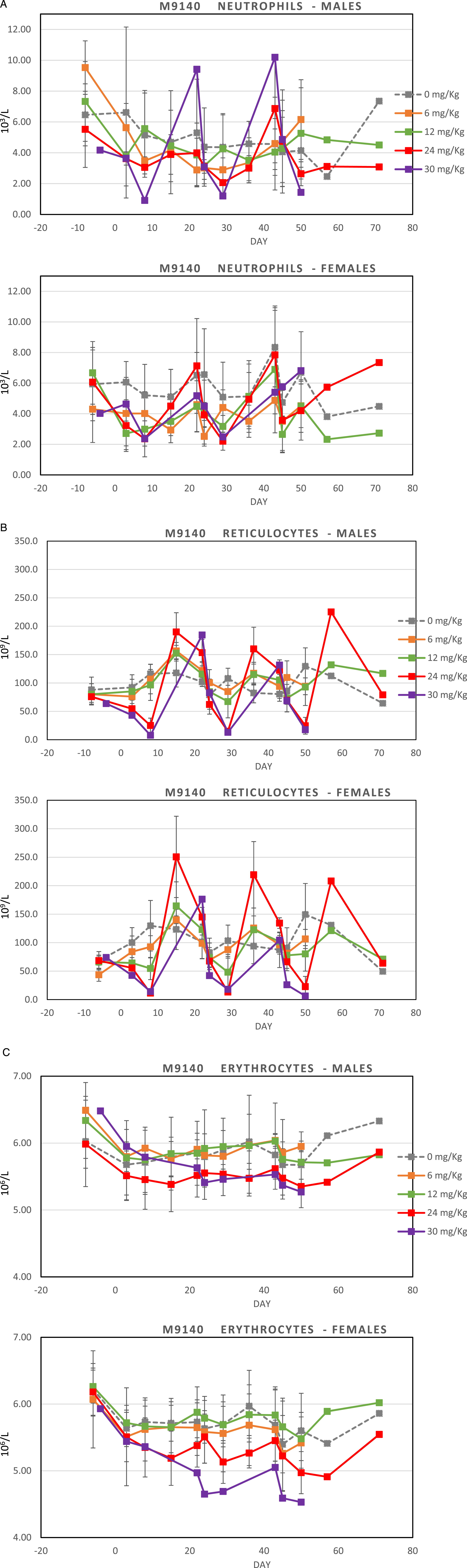
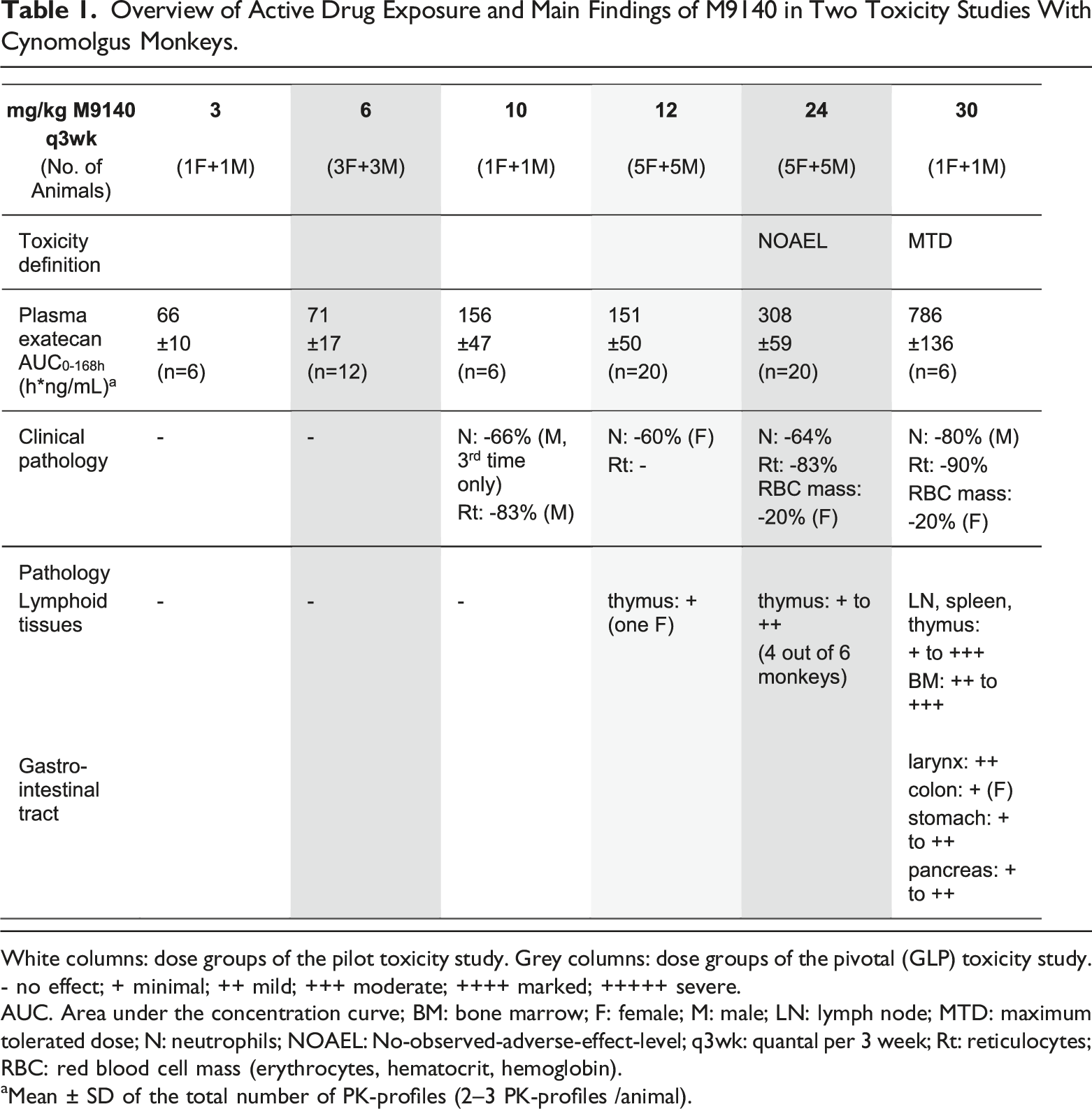
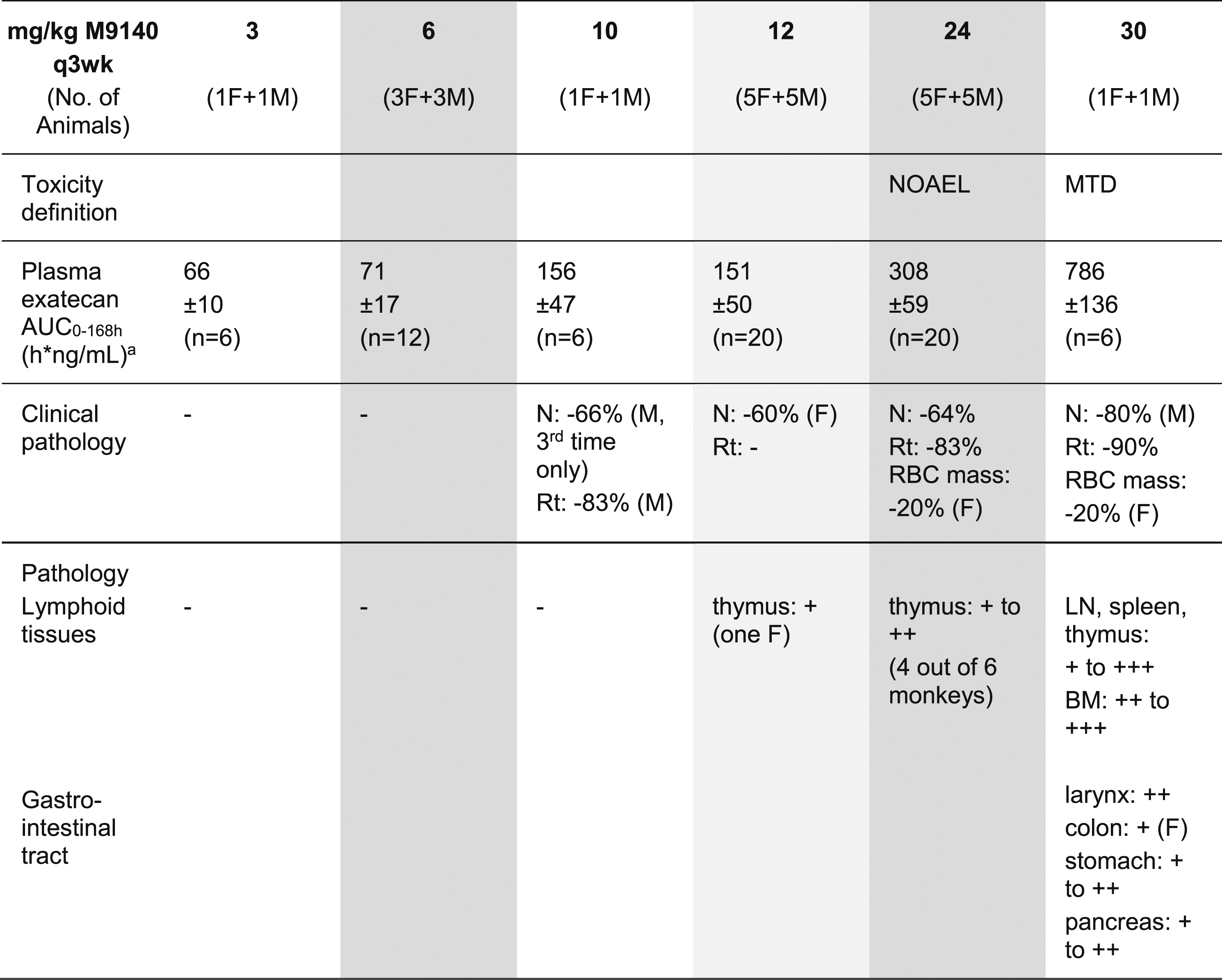
Microscopic examinations demonstrated at 30 mg/kg up to moderate and at 60 mg/kg up to severe changes of the hematolymphoid system consisting of decreased cellularity of the bone marrow (Figure 3A), decreased size and number of germinal centers and lymphoid depletion in thymus, spleen, and lymph nodes (Table 1). The Peyer’s patches showed a mild lymphoid depletion in the male at 60 mg/kg. Alterations of the digestive tract were slight at 30 mg/kg and up to severe at 60 mg/kg. At 30 mg/kg, minimal mucosal atrophy, crypt necrosis, and glandular dilation in the colon (only female, Figure 3B) were present as well as up to mild atrophy in the stomach, the exocrine pancreas (acinar cells), and the larynx (glottis epithelium). At 60 mg/kg, compared to the previous dose level, these locations and the small intestine and esophagus were increasingly adversely affected. Also, moderate atrophy in the salivary gland was noted. No lung changes (Figure 3C) were observed at any dose, and no histopathological changes were noted at 3 and 10 mg/kg per occasion. Key histopathological changes after repeat-dosing of M9140 to monkeys. (A) Microscopy of the bone marrow. (B) Microscopy of the colon. (C) Microscopy of the lung. Representative pictures of hematoxylin and eosin-stained slides of the bone marrow (A) showing decreased cellularity compared to untreated control animal (insert), colon (B) with minimal crypt necrosis, glandular dilatation (arrows) and mucosal atrophy, and normal lung (C) 1 week (D50) after repeated dosing (3x) of 30 mg/kg M9140 once every 3 weeks to cynomolgus monkeys. Overview of Active Drug Exposure and Main Findings of M9140 in Two Toxicity Studies With Cynomolgus Monkeys. White columns: dose groups of the pilot toxicity study. Grey columns: dose groups of the pivotal (GLP) toxicity study. - no effect; + minimal; ++ mild; +++ moderate; ++++ marked; +++++ severe. AUC. Area under the concentration curve; BM: bone marrow; F: female; M: male; LN: lymph node; MTD: maximum tolerated dose; N: neutrophils; NOAEL: No-observed-adverse-effect-level; q3wk: quantal per 3 week; Rt: reticulocytes; RBC: red blood cell mass (erythrocytes, hematocrit, hemoglobin). aMean ± SD of the total number of PK-profiles (2–3 PK-profiles /animal).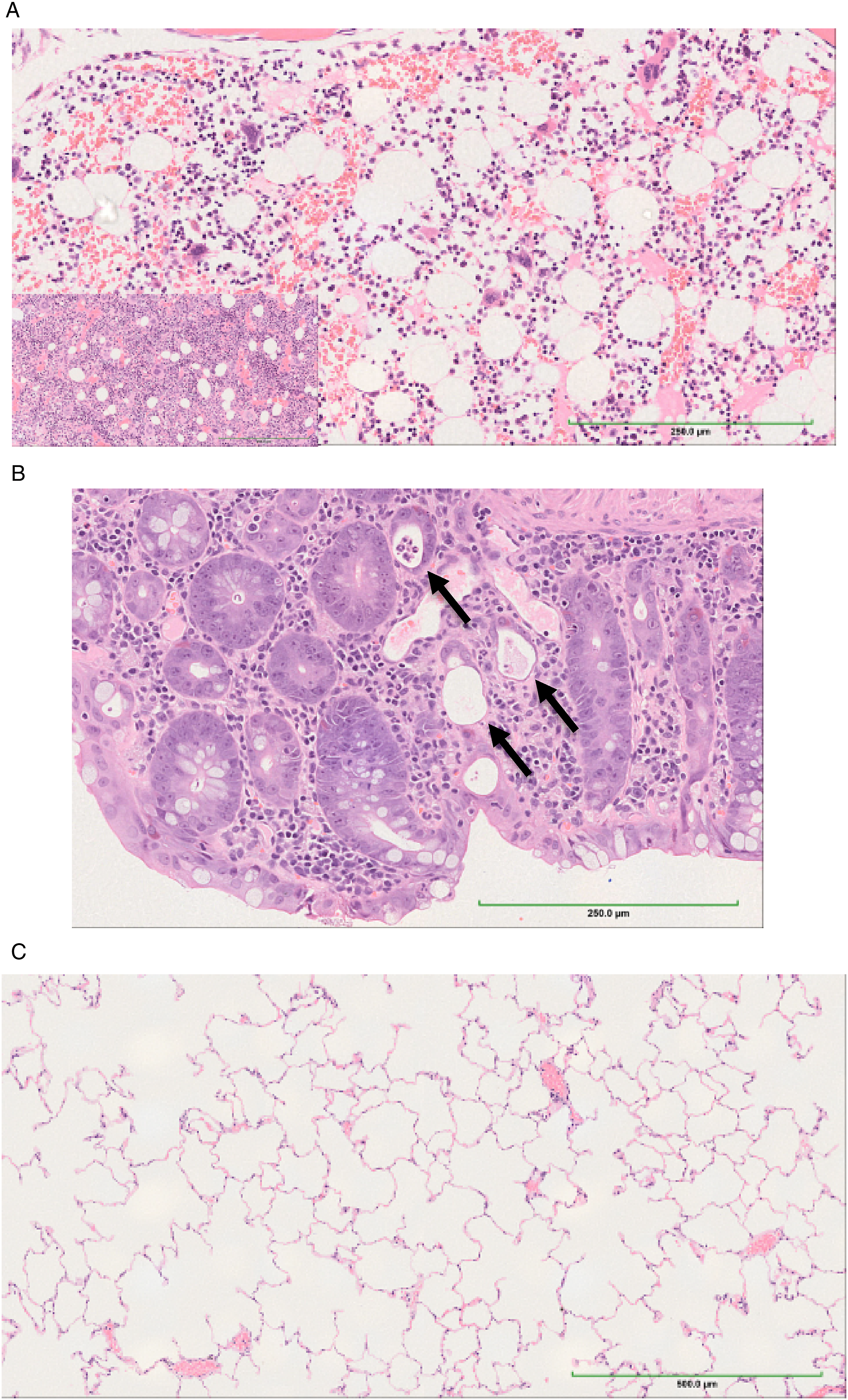
Pivotal 7-Week Toxicity Study
Due to the clinical adverse effects and BW-decrease in the female at 30 mg/kg (after the third infusion) in the pilot study, the highest dose of M9140 in the pivotal study was set at 24 mg/kg. No test item-related clinical signs, ophthalmology, or body weight changes were observed during treatment or recovery periods, except for one female treated at 12 mg/kg/occasion that showed a slight body weight decrease from Day 7 (−7.5% vs basal at the end of the dosing period) and reduced food intake from the second infusion onward.
At 24 mg/kg/occasion, clinical pathology investigations (hematology, chemical chemistry, immunophenotyping, coagulation and urinalysis) showed transient decreases in neutrophils (up to −64%) and reticulocytes (up to −83%) in both sexes 48 and 168 hours after each dosing occasion, followed by a rebound effect (Figures 2A and B). In addition, females showed decreased red blood cell mass of up to about −20% (Figure 2C). At 12 mg/kg/occasion, similar changes in neutrophils were observed in females only (up to −60%). No changes were seen at 6 mg/kg/occasion. These hematological changes are consistent with previously published effects of exatecan mesylate,7,23,30 causing toxicity on rapidly proliferative tissues, including hematopoietic tissues.
Repeated intravenous infusions of the test item M9140 did not result in toxicologically relevant alterations in organ weights (data not shown) or histopathology at any dose (Table 1).
Safety Pharmacology Assessment
In the pivotal toxicology study, electrocardiograms (ECG), heart rate, blood pressure, respiratory rate, body temperature, and clinical neurological investigations did not reveal any drug-related effects in the treated monkeys. Also, in the pilot study no test item related changes were noted on cardiovascular parameters of monkeys treated repeatedly up to 30 mg/kg of M9140. Body temperature recordings were generally in the physiological range with two exceptions. In the single monkey infused with 60 mg/kg on one occasion, the body temperature was reduced to 35.0°C on the day of preterm sacrifice (day 9), and hypothermia (36.1°C) was observed on the day of scheduled sacrifice (day 50) in the female monkey treated at 30 mg/kg.
Overall Conclusion Toxicity Studies
Since the transient hematological effects in the pivotal toxicity study was within normal laboratory reference limits and occurred in the absence of any relevant histopathological changes or clinical signs, these changes were not considered adverse equating the no observed adverse effect level (NOAEL) at 24 mg/kg q3wk. The cyclic reductions of neutrophils and reticulocytes after each dose (with nadir of 7 days) and regenerative anemia observed during the study (in females) were attributed to the pharmacological effects of low concentrations of released exatecan from the ADC. The maximum tolerated dose (MTD) was considered 30 mg/kg q3w mainly due to the observed adverse clinical signs and BW-decrease in the female only which appeared at the end of the study (after the third infusion). The main target organs of toxicity identified was restricted to the GI and the hematolymphoid system.
Toxicokinetic Evaluation in Toxicity Studies
Toxicokinetic Evaluation in the Pilot Toxicity Study
On all sampling days the exposure to all M9140 analytes (total Ab, conjugated exatecan, and unconjugated exatecan) increased roughly proportionally to the increasing dose from 3 to 30 mg/kg/occasion (Days 1, 22, and 43). At 60 mg/kg, despite proportional increase of total Ab and conjugated exatecan, unconjugated exatecan exposure (AUC0–168 h) in the single male was disproportionally increased, about 10-fold higher than at 30 mg/kg. Plasma anti-drug antibody measurement did not reveal antibody formation against M9140 across the dose groups.
Toxicokinetic Evaluation in the Pivotal Toxicity Study
After 0.5 h intravenous infusion, on Day 1 (first administration) and Day 43 (third administration), the peak concentration (Cmax) for both conjugated and total antibody was reached 0.5–2 h from the start of infusion in all dose groups. Conjugated and total antibody plasma concentration decreased generally with a biphasic profile, with no differences in the shape of the curve among dose groups. After the first administration, the mean terminal half-life (t1/2), calculated for conjugated and total antibody, was similar in all dose groups ranging from 106 to 110 h in males and 96.4 to 125 h in females for conjugated antibody, and 100 to 104 h in males and 90.8 to 115 h in females for total antibody.
After both administrations, the peak concentration (Cmax) of unconjugated exatecan was reached later than that of the conjugated antibody; the tmax ranging from 2.0 to 6.0 h from the start of infusion in all dose groups. The plasma concentration vs time profiles of unconjugated exatecan decreased faster than conjugated antibody with quantifiable levels up to 96–336 h. Terminal t1/2 calculated for unconjugated exatecan ranged from 53.0 to 60.2 h in males and 35.1 to 65.2 h in females.
On all sampling days, the exposure [Cmax, AUC0–504 h (Day 1) and AUC0–168 h (Day 43)] to conjugated antibody, total antibody, and unconjugated exatecan in plasma increased roughly proportionally to the increasing dose from 6 to 24 mg/kg/occasion in males and females. After repeated administrations on Day 43, the exposure to all analytes was comparable to Day 1, with no accumulation or drop in exposure observed in any dose groups. No sex differences in plasma exposure to any analyte were observed in any dose group.
No remarkable variation in individual exposure were observed at any dose of pharmacokinetic (PK) investigations, and no indication for immune-complex mediated toxicity was found suggesting no interference of exposure by anti-drug antibody formation. Consequently, no analysis of anti-drug antibodies was performed.
Toxicokinetic Evaluation Across the two Toxicology Studies
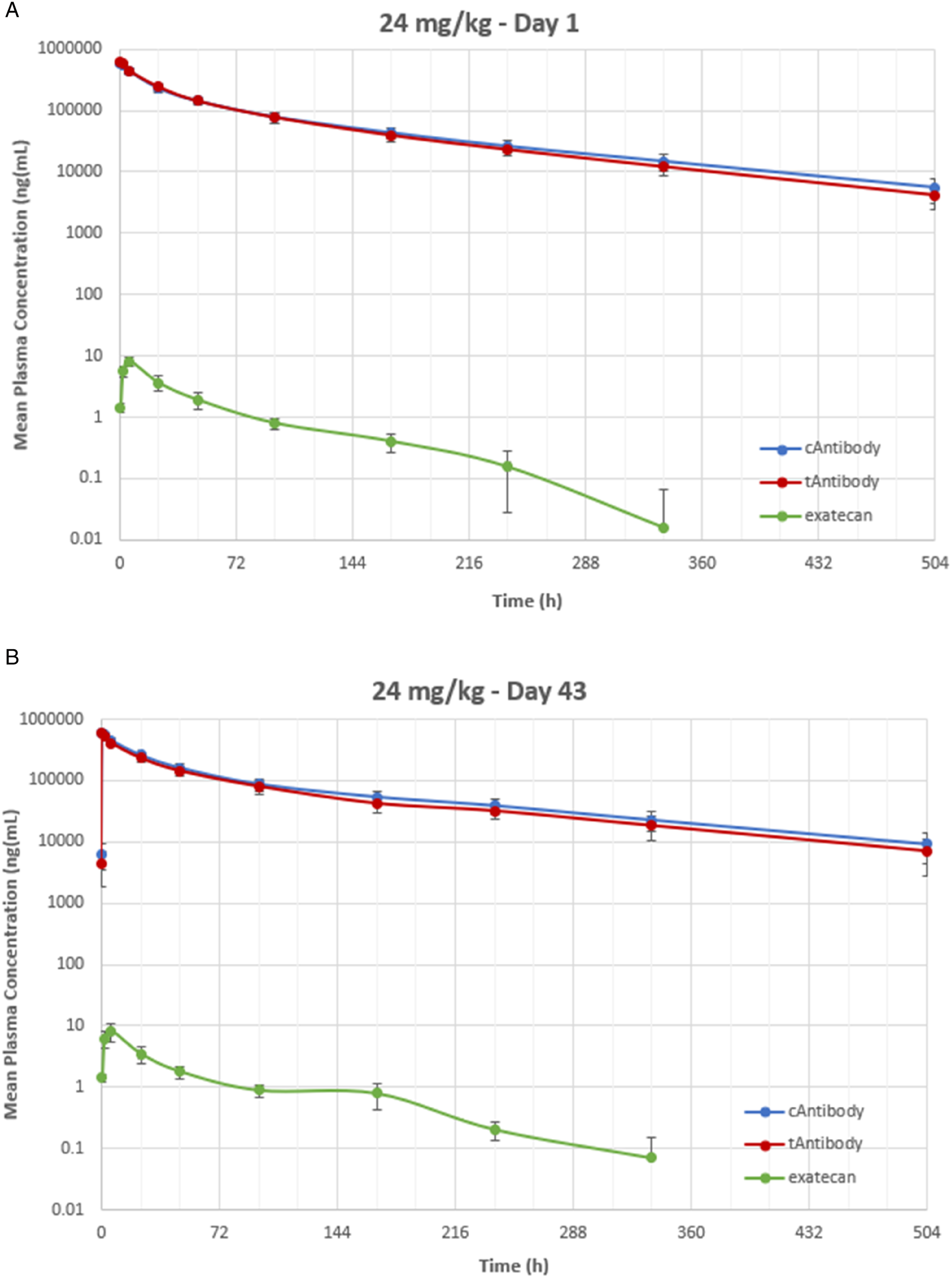
To allow comparison of dose- and exposure-response relationship across the two independent studies with an identical repeat-dosing schedule (3x q3w) and using the same origin of Vietnamese cynomolgus monkeys and test facility, total antibody concentrations in plasma (measured by identical immunoassay method) were taken at the end of the 30-minute i.v. infusion (Cmax in µg/mL) to analyze infusion consistency and dose-proportionality across the applied M9140 doses. The i.v. infusion across the 2 studies was consistent (Figure 4A) with somewhat higher infusion at 3 mg/kg and at 60 mg/kg (one animal: total Ab/dose = 34.7, not depicted in the Fig) resulting in a dose-proportional increase of total Ab exposure in plasma (Figure 4B). Since total Ab exposure and conjugated Ab plasma exposure were almost superimposable on the pivotal toxicology study and the amount of released exatecan was low, both parameters can be regarded as a reflection of the ADC exposure in plasma (Figure 5). About 80%–90% of exatecan release from the ADC occurs over the first week (AUC0–168 h vs AUC0–504 h). (A) Intravenous infusion consistency across the two toxicity studies in monkeys. (B) Dose-proportional infusion across the two toxicity studies in monkeys. Mean plasma total antibody (tAb)/dose (A) and mean ± SD of total Ab (B) immediately following infusion (Cmax) of 6, 12, and 24 mg/kg M9140 in the pivotal GLP study and 3, 10, and 30 mg/kg M9140 in the pilot study with cynomolgus monkeys. The group size was 1/sex at 30 mg/kg, 3/sex at 6 mg/kg, and 5/sex at 12, and 24 mg/kg. Typical toxicokinetic profile with three analytes in plasma of monkeys. Typical example of TK profiles following 24 mg/kg infusion of M9140 showing highly comparable conjugated (c) Ab and total (t) Ab concentrations and very low unconjugated exatecan concentrations in monkey plasma at day 1 and day 43. The group size was 5 monkeys/sex at 24 mg/kg.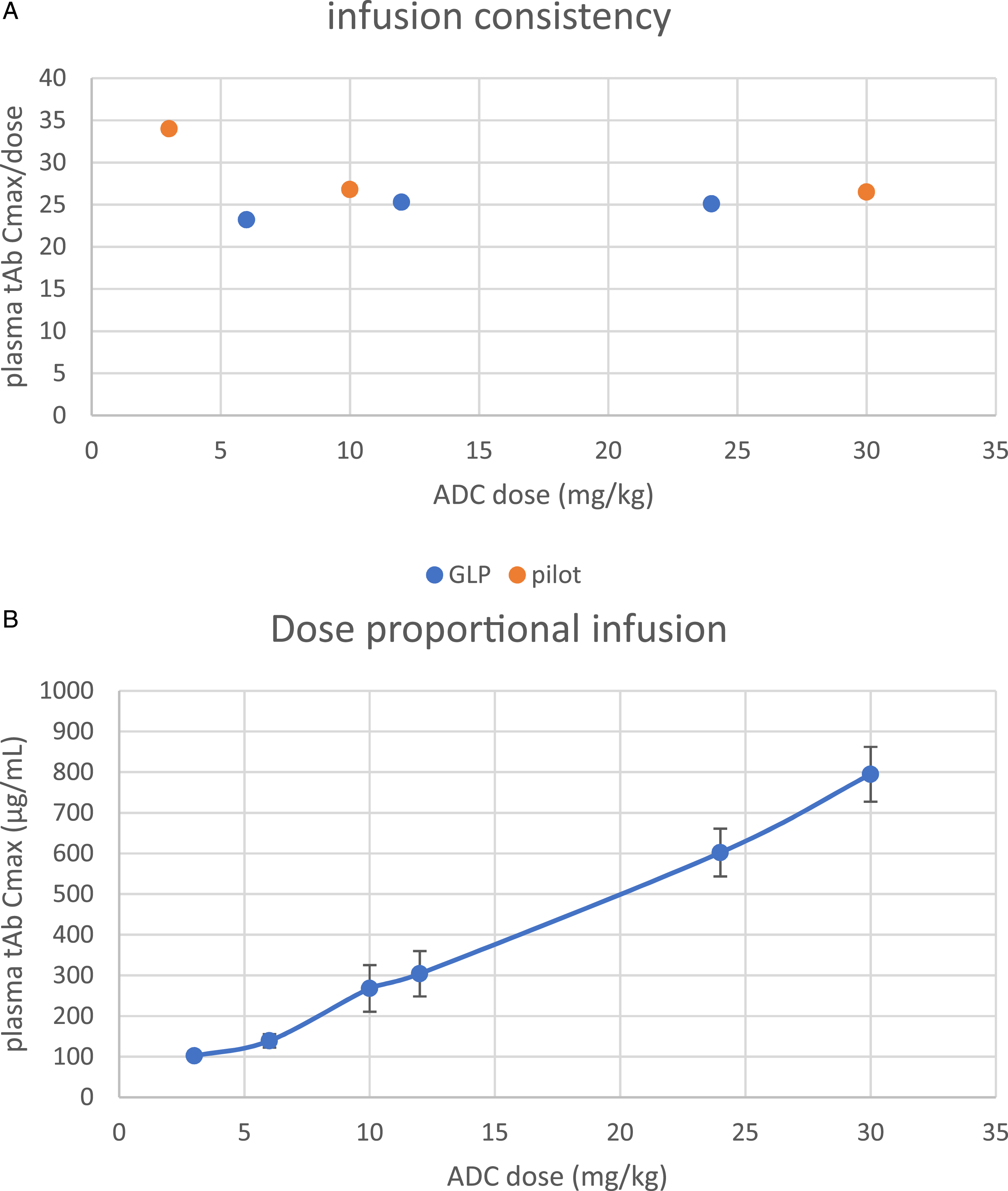
The low concentrations of unconjugated exatecan gradually released from the ADC were measured in plasma (range of about 0.1–10 ng/mL) and increased proportionally with increasing ADC dose or with the plasma total Ab concentrations at the end of infusion except for the higher doses of 30 mg/kg and 60 mg/kg M9140. The small dose increment of 1.25-fold from 24 to 30 mg/kg resulted in about 2.5-fold difference in plasma exatecan (AUC0–168 h) either expressed against the M9140 dose (Figure 6A) or plasma exposure of total Ab immediately after the infusion (Figure 6B) or total Ab exposure (AUC0–168 h)(Figure 6C). This super-proportional increase was much more pronounced at 60 mg/kg (∼26-fold vs the 2.5-fold lower dose of 24 mg/kg, not depicted). Proportionality of exatecan plasma exposure after M9140 infusion to monkeys across the two toxicity studies. Mean ± SD of plasma exposure of exatecan (AUC0–168 h) plotted against (A) the dose of M9140, (B) against peak plasma total Ab (mean Cmax ± SD) immediately after infusion, and (C) against plasma total Ab exposure (mean AUC0–168 h ± SD) across the two toxicity studies in monkeys. The group size was 1/sex at 30 mg/kg, 3/sex at 6 mg/kg, and 5/sex at 12, and 24 mg/kg.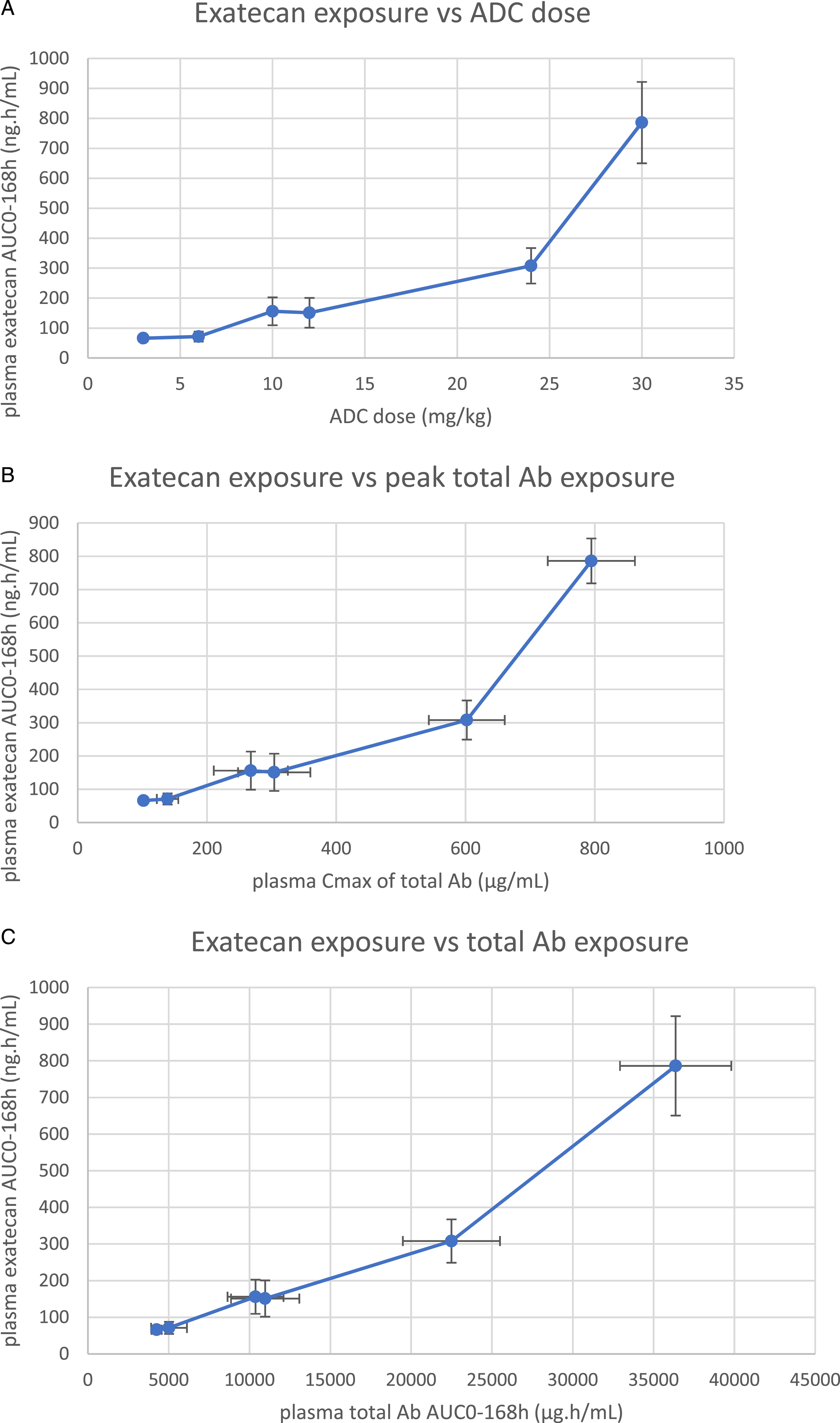
Interestingly, this evaluation across the two studies of released exatecan in plasma supports the hypothesis that the observed toxicities are due to the liberated exatecan as the main driver of toxicity, as summarized in Table 1. The exposure to liberated exatecan (AUC) likely explains the difference in toxicity for the relatively small dose increment (1.25-fold) of 24 to 30 mg/kg (no histopathology vs histopathological effects, respectively). The steep increase of toxicity at 60 mg/kg given once (not tolerated) vs 30 mg/kg q3w (2-fold dose factor) was associated with approximately 10-fold increase of plasma exatecan exposure (AUC0–168 h).
Overall, the data supports the NOAEL at 24 mg/kg (q3w) and the MTD at 30 mg/kg associated with about 2.5-fold difference in plasma exatecan exposure (AUC0–504 h) between the two high dose groups of each study (pilot: 884 ± 83.7 ng.h/mL, pivotal: 363 ± 47.9 ng.h/mL) that determined the dose-toxicity relationship.
Discussion
Repeated iv-infusion of M9140 to cynomolgus monkeys at 3 to 30 mg/kg/occasion for 3 times every 3 week (3x q3wk), induced dose-dependent adverse effects in the hematolymphoid and GI systems. The effects of M9140 can be mainly attributed to exatecan gradually released from the ADC at low ng/mL concentration but exerting potent antimitotic and cytotoxic properties. At 24 mg/kg (NOAEL) or higher, transient reductions in neutrophil and reticulocyte counts were observed with each dosing event and mild, reversible anemia was noted throughout the study (Figures 2A–C). Notably, no lung toxicity was observed after M9140 up to 30 mg/kg (MTD) in these repeat-dose toxicity studies that were similar in study design to those used to confirm ILD with T-DXd in cynomolgus monkeys. 9 Since the non-human primate model is regarded as predictive for the risk of ILD in patients, 11 this distinctive result with M9140 indicates a minimal risk for ILD when dosing patients with M9140 for the treatment of solid tumors overexpressing CEACAM5.
M9140 Safety Evaluation
The toxicity profile of M9140 obtained in monkeys resembles that of exatecan mesylate as was previously evaluated in rodents and dogs, as well as in several clinical trials with oncology patients.7,8,23 The toxicity profile of exatecan is highly consistent across all species targeting mainly the hematolymphoid system, with neutropenia as the principal dose-limiting toxicity, and the GI-tract. Rapidly proliferative tissues such as hematopoietic and lymphoid tissues (bone marrow, lymph nodes, associated with neutropenia, thrombocytopenia, anemia, lymphopenia), GI-mucosa (associated with vomiting, diarrhea), reproductive tissues, and hair follicles have been most prone to the toxic effects of exatecan mesylate. The observed toxic effects to hematolymphoid tissues by M9140 were attributed to the released exatecan solely because hematopoietic cells lack CEACAM5 expression.26,27 Most GI-tract effects of up to 30 mg/kg M9140 were mild and can be largely ascribed to the substantial increase of exatecan (2.5-fold) and its known toxicity to the digestive tract, particularly to the fast-dividing epithelial cells in the mucosa. Though the increase in Ab concentration was small from 24 to 30 mg/kg (+25%), it cannot be fully excluded that some GI-tract effects may be related to the contribution of payload delivered via the target as observed at the MTD of 30 mg/kg q3wk such as the minimal effects in the colon (see Figure 3B) and mild atrophy of the glottis epithelium of the larynx. As previously reported, CEACAM5-expressing epithelial tissues were located in the digestive system (larynx, salivary gland, esophagus, colon), upper respiratory system (trachea), uterus (glandular), and skin (glandular), but not for instance in hematolymphoid tissues (bone marrow, spleen, and lymph nodes) and the lung parenchyma.26–28 This was confirmed with 8G4SO Ab staining (the Ab backbone of M9140) from a panel of 32 tissue types of cynomolgus monkeys (unpublished data).
The similar kinetic profiles of conjugated Ab and total Ab in monkey plasma suggest adequate stability of the ADC with a half-life of about 4–5 days. Despite the relatively low plasma levels of exatecan liberated from the ADC (Cmax < 24 ng/mL at MTD) mostly over a 1-week period, the effects of M9140 were mainly attributed to exatecan exerting potent cytotoxic properties. At the non-tolerated dose of 60 mg/kg (dosed once), the observed increased severity of toxicity remained restricted to the GI-tract and hematolymphoid system, not affecting for instance the lung or other pivotal organs (day 9 necropsy). At this dose, also the esophagus expressing CEACAM5 was targeted. The increased toxicity observed at 60 mg/kg was associated with a stronger supra-proportional effect of unconjugated exatecan between 30 and 60 mg/kg (10-fold) presumably due to increasingly disturbed (metabolic) clearance of exatecan via cytochrome P450 3A4/1A2 enzymes. Since the two identified main hydroxylated metabolites of exatecan (4-hydroxymethyl- and 3-hydroxy-exatecan) were substantially less cytotoxic than exatecan,7,8 it appears to represent a key detoxifying pathway. In this regard, the relatively benign effects in the intestine as compared to the hematolymphoid tissues at 30 mg/kg may be due to substantial CYP3A metabolism of exatecan in this compartment. Recently, the declared clinical MTD of M9140 in the escalating part of the study was 2.8 mg/kg in 3-week courses showing mostly hematological adverse events, while GI-related adverse events were of low grade 29 corroborating monkey safety data. The relatively large discrepancy in the established MTD between monkeys (30 mg/kg) and patients (2.8 mg/kg) cannot be deduced by simple allometric scaling by body surface area (dividing animal dose by conversion factor 3.1 to yield the human equivalent dose: 9.7 mg/kg) 33 but may be best explained by the at least 10-fold faster clearance of exatecan in monkeys (t1/2 < 1 h, unpublished data) compared to patients (t1/2 : 9–12 h) 8 when dosed with exatecan. The exposure to exatecan depends on the relatively slow release from the ADC which is roughly similar in monkeys (t1/2 : 2–3 days) and patients (t1/2:5 days). However, ultimately the liberated exatecan is cleared more than 10-fold faster in monkeys leading to comparable exposure despite the ca. 10-fold M9140 dose difference. In other words, the monkey eliminates exatecan much more efficient than human independent of its drug presentation resulting in the higher tolerability in monkeys.
What Is the Putative Mechanism Behind ILD?
In a parallel program using the same anti-CEACAM5 Ab (as in M9140) but different linker (tripeptide) to conjugate exatecan, a similar toxicity profile as for M9140 was noted without any lung findings in a similarly designed pilot toxicity study as was conducted for M9140 (unpublished data). In another ADC program using the identical linker-payload construct (as for M9140) but different Ab targeting GD2, no lung findings were noted in repeat-dose toxicity studies with monkeys. 30 Together, our exatecan-based ADC data, and data on camptothecin payloads alone (i.e., DXd, SN38) and other antibody camptothecin-conjugates (as described in the Introduction) are suggesting ILD is both target- and payload-independent and allude to an apparent unique property of DXd-based ADCs mediating ILD as observed with several assets of the same ADC platform technology.4,13,15 A hypothesis on the possible mechanism of ILD was devised as follows.
The antibody and payload of T-DXd are linked by a maleimidocaproyl(mc)-containing tetrapeptide (-GGFG-) with an aminomethylene (-NH-CH2-) spacer attached to the 2-oxyacetyl (-O-CH2-C(=O)-) amino-group of DXd (T-mc-GGFG-aminomethylene-DXd), that can be enzymatically cleaved between the last glycine and aminomethylene moiety (synonym: methylamine). After ADC internalization and lysosomal protease cleavage, a temporary H2NCH2O-DXd intermediate is formed and methylamine is rapidly hydrolyzed to ammonia and formaldehyde by design of the self-immolative spacer according to Nakada et al.,
22
leaving DXd (2-hydroxyacetyl-exatecan) (Figure 7). Catabolism of trastuzumab-deruxtecan and proposed induction of ILD. Proposed release mechanism of deruxtecan (DXd) from Trastuzumab-mcGGFG-MA-deruxtecan concomitantly with methylamine (MA) producing additional toxic by-products (formaldehyde, ammonia)
22
and proposed local AO-processing of methylamine as substrate further generating toxic by-products (such as aldehydes, ammonia, and hydrogen peroxide), oxidative stress and inflammatory responses, that may play a role in the formation of ILD. Green boxes display the features of ILD in monkeys as described by Kumagai et al.
11
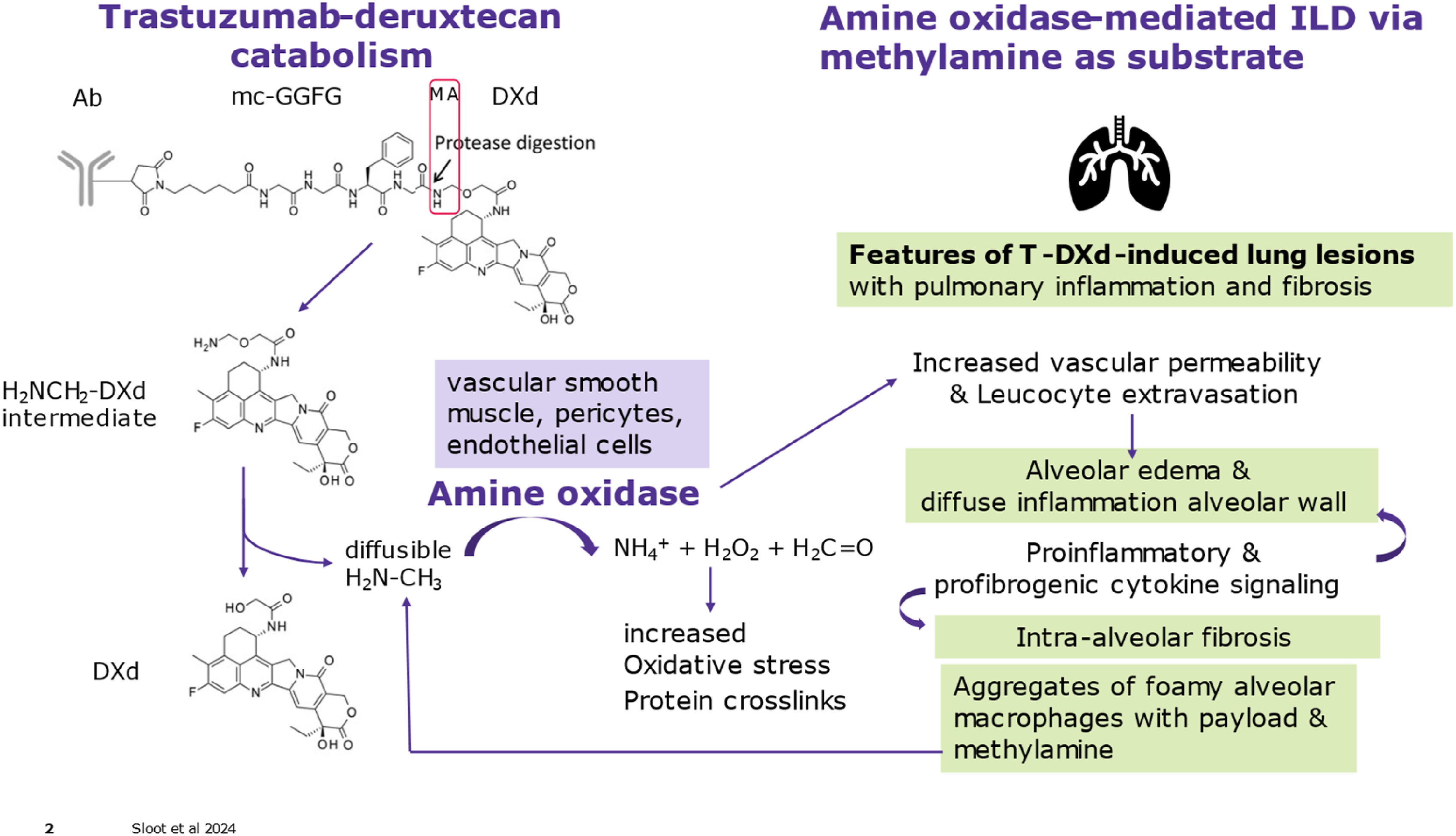
However, it is conceivable that methylamine may diffuse locally and induce toxic by-products by another process that may reach adverse levels under some conditions. Notably, mechanisms involving enzymatic deamination of methylamine by semicarbazide-sensitive amino oxidases (SSAO) such as the family member vascular adhesion protein 1 (VAP-1) produces toxic formaldehyde, ammonia, and hydrogen peroxide which under specific conditions such as disease, drugs or toxins including methylamine, oxidative stress, lipopolysaccharides (LPS), or cigarette smoke can ultimately lead to strikingly similar pulmonal fibrotic and inflammatory reactions31,33 as described for T-DXd-induced ILD (see Introduction). The amine oxidase (AO) enzymes capable of short-chain deamination such as methylamine (specifically by AO3) are located on intracellular vesicles as well as on the cell surface of vascular smooth muscle, adipocytes, pericytes, and endothelial cells, and the human lung is exhibiting substantial SSAO activity, higher than for instance in rodent lungs.31–33 Once triggered by liberated methylamine and mediated by toxic by-products from AO-activity, local vascular changes (e.g., increased permeability, alveolar edema) and extra/intracellular damage and deposits such as protein cross-linkage and oxidative stress may be slowly built up and aggravated by local inflammatory reactions (e.g., cytokine responses/signaling) from infiltrating leucocytes/monocytes, ending up in fibrosis. In this process, the enzymatic activity of SSAO (VAP-1) is required for leucocyte extravasation through the endothelium (e.g., microcapillary vessels). In addition, immunostaining of DXd to localize T-DXd distribution at necropsy timepoints of 1.5 or 3 months in monkeys revealed target-independent uptake of T-DXd in alveolar macrophages and liver macrophages. 11 Perhaps, the distribution of T-DXd to alveolar macrophages and high AO-activity in human pulmonary tissue may explain the sensitivity for ILD in a proportion of humans (and monkeys), since no lung effects were found in 1.5-month rat toxicity studies with T-DXd. 9 The increased incidence of ILD and infiltration of lymphocytes in thickened, fibrotic alveolar walls after 3 months compared to 1.5 month in monkeys dosed with T-DXd 11 is indicative of the relatively slow, accumulative nature of the pathological process suggested above. In addition, transgenic mice overexpressing human VAP-1 (SSAO) are vulnerable to LPS and inhaled methylamine toxicity. 31 Together, it is postulated that the co-release of methylamine and toxic by-products (formaldehyde, ammonia) from GGFG-methylamine-DXd-containing ADCs and local AO-processing of methylamine as substrate may further generate toxic by-products (such as aldehydes, ammonia, and hydrogen peroxide), oxidative stress and inflammatory responses, that may play a role in the formation of ILD. Insights into the mechanism of ILD may contribute to possible treatment of such effects or prevent ILD by designing different drug-linker constructions without toxic spacer.
In this respect, M9140 composed of a hydrophilic, enzymatically cleavable maleimide-containing β-glucuronide linker to release exatecan after internalization and lysosomal uptake without the possibility of generating any methylamine-related toxic by-products (Figure 1).
Conclusion
The adverse effects of M9140 obtained in non-human primates were confined to GI and hematolymphoid tissues and was driven mainly by antigen-independent toxicity of the liberated payload. In the absence of any lung effects in 1.5-month toxicity studies with M9140 in monkeys, no relevant risk for ILD is expected when dosing M9140 to patients for the treatment of solid tumors overexpressing CEACAM5 such as CRC and GC (ClinicalTrials.gov Identifier: NCT05464030). Moreover, there is no intrinsic potential for M9140 to induce ILD as postulated for the unique GGFG-methylamine-DXd-based ADCs from the hypothetical release of methylamine and proposed toxic by-products directly and/or via local AO-processing of methylamine as substrate. At present, no event of ILD has occurred in over 40 patients dosed with M9140 who were exhibiting mostly hematological side effects and relatively benign GI-tract events 29 as predicted by the non-human primate safety studies. The nonclinical benefit-risk profile of M9140 as targeted oncology therapy is anticipated to provide an acceptable clinical therapeutic window with manageable and predictable safety risks.
Footnotes
Acknowledgments
The authors would like to thank the following Merck Healthcare KGaA employees: Jan Anderl, Nir Berger*, Michela Carbonatto, Valeria Castagna, Stephan Hecht, Seema Kumar*, Luca Marchiando, Rita Mastroianni, Federico Sirtori, Michael Schmitt, Stephan Schmidbauer*, Matthias Winzer, for all their dedicated work, and Eric van Esch (InSight Pathology) for contracted pathology evaluation of the pivotal toxicity study. Special thanks go to Jan Anderl and Nicole Huebler for critically reviewing the manuscript.*) former employee of (a business of) Merck KGaA, Darmstadt, Germany
Author Contributions
The authors and co-workers on the studies co-designed the experiments, collected, analyzed, interpreted, and reported the data. The key author WS wrote the manuscript and devised the ILD hypothesis, and decided to submit the article for publication, and all authors reviewed and critically evaluated and approved the manuscript to ensure accountability for data accuracy and integrity.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors are employees of Merck KGaA and may own stock in Merck KGaA. WS has (pending) patent rights on M9140 related materials.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: All studies were funded by Merck Healthcare KGaA.