
Abstract
The skin is the largest organ in the body and the only one to come into contact with solar UV radiation (UVR). UVA (320-400 nm) is a significant contributor to UV-related skin damage. The UVA spectrum makes up over 95% of solar-UV energy reaching the earth’s surface causing the majority of the visible signs of skin photoaging. Many consumer products also emit UVA, including nail dryers. There have been sporadic reports suggesting that these units may be contributing to skin cancer incidence. This notion was recently bolstered by a finding that nail dryer-irradiated mammalian skin cells develop a mutational signature consistent with UVA exposure. This report was surprising considering the comparatively low level of UVA to which the skin is exposed during nail treatments. In this research, we investigated how UVA-emitting devices caused cytotoxic/genotoxic impact after only low levels of UVA exposure. Our data showed that levels of UVA in the unit are highly variable and location dependent. We confirm previous reports that using prolonged exposure protocols could induce significant levels of DNA damage. It was also determined that UV-induced DNA damage only partially correlated with the level of UVA fluency. On investigation, we found that the unit had a rapid increase in internal temperature when in use. Exposing human cells to these elevated temperatures acted synergistically with UVA to magnify the cytotoxic and genotoxic impact of UV irradiation.
Keywords
Introduction
UVA radiation can induce the generation of reactive oxygen species (ROS) such as singlet oxygen (O2), superoxide anions (O2• -), and hydrogen peroxide (H2O2), which can damage DNA by oxidizing the DNA bases. Guanine (G) is particularly susceptible to this oxidation by ROS due to its low oxidation potential. This breakdown leads to the formation of 8-oxo-7,8-dihydro-2’-deoxyguanosine (8-oxo-dG). 1 8-Oxo-dG causes guanine-to-thymine mutations. UVA radiation has also been found to induce DNA damage via cyclobutane pyrimidine dimers (CPDs), which are linked to the development of melanoma and other skin cancers. This has been proposed to occur when ROS and nitrogen species generated by UVA excite electrons in melanin, leading to the transfer of energy to DNA and the formation of CPDs. This process continues to occur long after UV exposure ends. 2 Oxidative damage caused by UVA has an impact on the dermal layer of the skin, leading to alterations in the perinuclear endoplasmic reticulum subdomain, 3 as well as changes in the structure of the extracellular matrix. 4 Further, this damage contributes to the photoaging of the dermis by the depletion of antioxidant enzymes, 5 characterized by wrinkles and loss of skin tone. 6
The use of UVA in the consumer market has been prevalent for years, notably in tanning beds and nail dryers. UVA nail dryers are devices used in salons, designed to cure gel nail polish quickly. While the use of these devices is brief, there is speculation around the overall safety of the units and whether they increase the risk of skin cancer.
7
Studies investigating the UV emission of these devices have reported that the spectral distribution and intensity varied substantially among devices, with some generating greater UVA radiation intensity (i.e., flux density) than that of natural sunlight. More specifically, a study that measured the output of 17 different UV nail dryers found that the amount of UVA emitted from the nail dryers varied considerably and calculated that the median UVA dosage (fluence) from the devices in a single session was 5.1 J/cm2, compared to the calculated threshold value for DNA damage of 60 J/cm2,
8
indicating that the median number of visits to even reach that threshold was 11.8. Many of these previously published reports did not use the nail dryer units according to the manufacturer’s specifications. A recent study by Zhivagui et al.
9
evaluated UV nail dryer-induced DNA damage and mutagenicity in a 2D cellular system. As the authors acknowledge, most salon sessions involve considerably less than 10 min of exposure using the nail dryers, yet the exposure protocol used in this study was either two 20-min exposures separated by an hour (acute exposure), or 20-min exposures on three consecutive days (chronic exposure; Figure 1).
9
This pattern of UVA exposure is unlikely to be encountered in a salon setting. Compared to typical use salon exposure, the acute/chronic cell radiation protocols used by this study delivered high UVA fluences of 18 to 27 J/cm2. However, we believe these studies fail to rigorously support the role of nail dryers in increased skin cancer risk and highlight the need for more accurate biological assays of genotoxic and cytotoxic damage induced by UVA from these devices. In the presented research, we compare the cytotoxic and genotoxic endpoints induced by normal-use salon exposure and excessive exposure UVA protocols. To this end, we used a high-throughput UVA irradiation unit to determine if it was possible to correlate genotoxic/cytotoxic damage from known amounts of UVA exposures to output from the nail dryers. During the measurements, we determined that the internal temperature of the nail dryer unit caused artefactually high levels of cytotoxicity/genotoxicity. These previously undescribed additional factors involved in the genotoxic impact of these machines largely preclude any results derived using 2D-cell culture models, from being used to guide human health decisions. Schematic of the exposure parameters of the three different protocols. Acute exposure is 2 × 20-min exposures with 60-min recovery between treatments. Chronic exposure is defined as 3 × 20-min exposure with 24 hr between treatments. Salon (real) exposure was designed to mimic the average single customer exposure during use in a salon; 6 × 30 sec exposures with 2 min between treatments.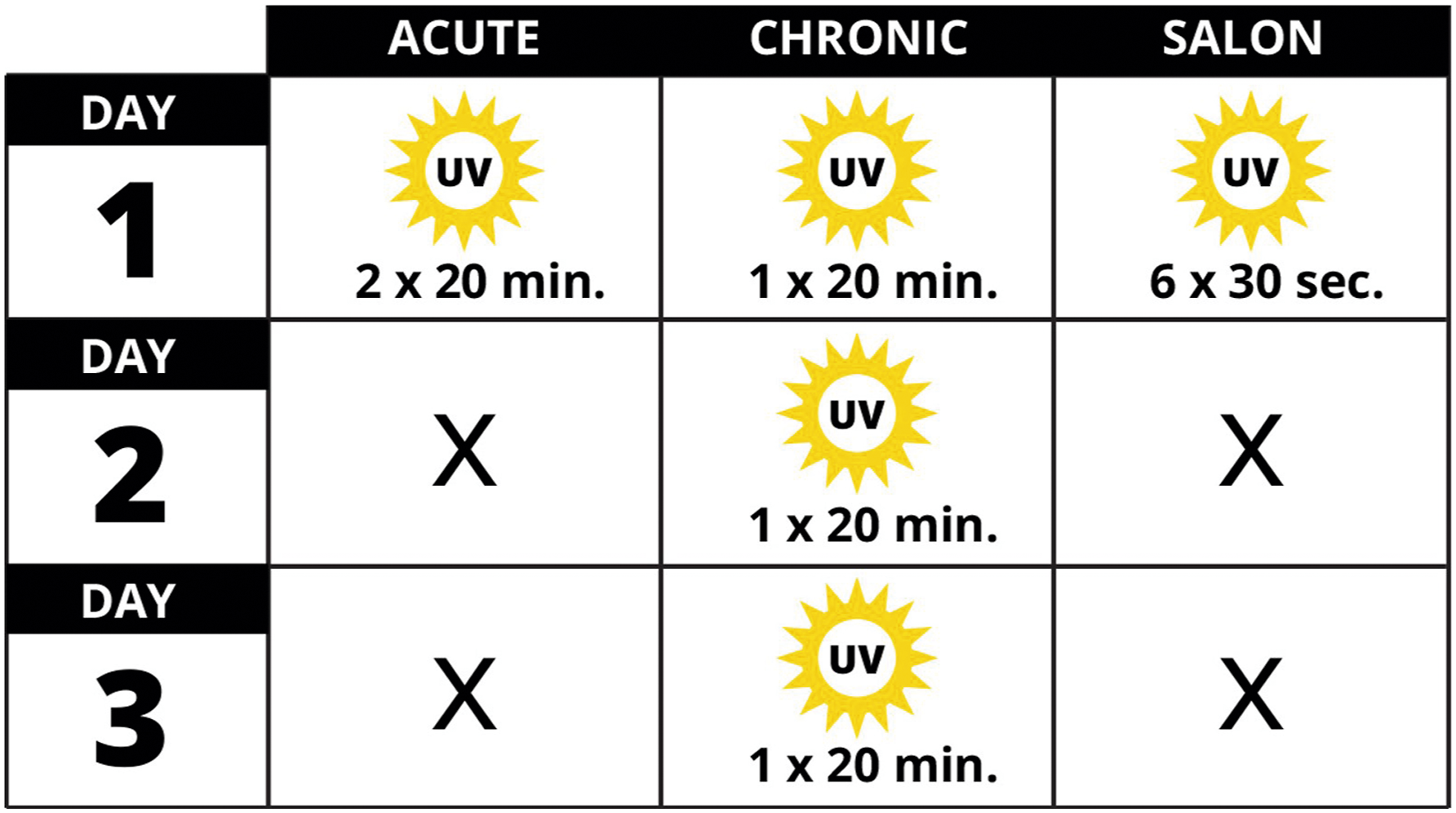
Materials and Methods
Cell Culture
E6/E7 immortalized Human Foreskin Keratinocytes (E6/E7-HFK) were grown and maintained in keratinocyte serum-free media (KSFM 1X, Gibco) supplemented with 25 mg Bovine Pituitary Extract (BPE), 2.5 µg Human Recombinant EGF, and 1% Penicillin/Streptomycin (PenStrep, Gibco). Immortalized Normal Human Dermal Fibroblasts (NHDF) were grown and maintained in Dulbecco’s Modified Eagle Medium (DMEM, Gibco), supplemented with 10% Fetal Bovine Serum (FBS, Peak Serum) and 1% PenStrep. All cells were cultured at 37°C in a 5% CO2 incubator.
UV Exposure Parameters
A 54W MelodySusie (MS) UV nail dryer (MelodySusie, Model: DR-6332B EOS9, Newark, CA) was used to determine the impact that commercial UV nail dryers have on the skin. This model of UV nail dryer houses 30 UV-LED lights that emit UVA light at 365 nm. As a comparative control, the LED UVR DNA damage induction system (LUDIS) was used with output calibrated using a microphotometer (International Light Technologies, Peabody, MA). The LUDIS is a high-throughput light system that allows for rapid and precise UVA (365 nm) exposure to cells in a 96-well format. The LUDIS platform was developed originally as a novel method to test compounds that may have utility as new sunscreen active ingredients. The use of UV-emitting high-powered LED lights is a major hardware innovation of the platform. Current UV-delivering devices (e.g., Philips TL-40 UVB; 40 J/m2s) are cumbersome with no ability to be used in high-through-put applications, having been in operation with essentially no major improvements for decades. Commonly used sources, such as mercury or neon lamps, are prone to wide-bandwidth UV output, making it particularly difficult to discern the relative contribution of a wavelength (UVA2 vs. UVA1) to inducing specific forms of DNA damage. Recent advances in LED technology have created microbulbs (like the lights that are used in the LUDIS) with very high light output with low heat induction emitting a narrow, very stable wavelength. UVA1 fluence must have physiological relevance to typical human exposure. While UVA irradiance fluctuates with weather, seasons, and time of day, roughly 5% of the 1000 J/m2 terrestrial solar radiation is UVR, of which 70-75% is within the UVA1 waveband. Thus, “typical” solar UVA1 irradiance (or flux density, sometimes thought of as “intensity”) is roughly 37.5 W/m2 (=1000 J/m2 x 5% × 75%). Thus, our highest fluence “dose” of 14.2 J/m2 represents a level of irradiation achievable in approximately one-hour (=142,000 J/m2
A major component of this research was to test different experimental parameters. The 3 exposure parameters used in this study were termed Acute, 9 Chronic, 9 and Salon. Acute exposure was 2 × 20-min UVA exposures with a one-hour recovery between treatments. Chronic exposure was 3 × 20-min UVA exposure with 24-hr recovery between treatments. Salon exposure was designed to simulate real exposure to UV nail dryers found in salons and was 6 × 30 sec exposures, with 2-min recovery between treatments.
Cell Viability and Cytotoxicity Assays
Cells were seeded in 96-well plates at 104 cells/well and were left overnight in a 37°C incubator prior to UVA exposure. Cells were then exposed to Acute, Chronic, or Salon UVA exposure as described. Viability measurements were conducted 48 hr after final exposure; fibroblast viability was measured using an XTT Cell Viability Assay Kit (Biotium, Fremont, CA, USA). Plates were incubated at 37°C for 4 hr, and absorbance was read at 450 nm using the BioTek Cytation7 cell imaging multimode reader. Keratinocyte viability was measured using DAPI stain after cells had been fixed with 4% paraformaldehyde and permeated with 0.2% Triton. Plates were incubated for 5 min at 4°C, and cell count was taken using the BioTek Cytation7 cell imaging multimode reader.
Assessment of Superoxide Formation with Dihydroethidium
To measure ROS accumulation in the cells exposed to UVA, Dihydroethidium (DHE) was used. 10 Cells were seeded at roughly 70000 cells per well in a black-sided, optical-bottom 96-well cell culture plate and were left overnight. Working concentrations of N-acetylcysteine (NAC) (2 mM), Antimycin A (50 µM), and DHE (10 µM) were prepared in phenol- and serum-free media. DHE alone was used to measure endogenous ROS, Antimycin A was used as a positive control, and NAC was used as a negative control. DHE at 10 µM was added to both the positive and negative control solutions. All media was removed, and cells were washed with 100 µL of pre-warmed serum- and phenol-free media before all working reagents were applied and placed in a 37°C incubator for 20 min. After incubation, a preliminary ROS reading was taken prior to cells being exposed to 0-14.2 J/cm2 UVA. A final ROS reading was taken immediately after UVA exposure. Endogenous ROS was subtracted from UVA-induced ROS in order to accurately visualize the amount of ROS induced by UVA.
Assessment of DNA Damage Using the Alkaline Comet Assay
To measure the amount of DNA damage accumulated in the cells after UVA exposure, the cells were run in an alkaline comet assay. 11 Cells were seeded in 96-well plates at 104 cells/well and were left overnight in a 37°C incubator prior to UVA exposure. Cells were then exposed to Acute, Chronic, or Salon UVA exposure as described. Immediately after the final treatment of UVA, cells were gravity-loaded into the comet chips (Biotechne, Minneapolis, MN) for 15 min. After loading, the comet chip was washed with PBS and sealed with 0.8% low-melting-point agarose (LMPA) before being placed in pH 10 lysis buffer solution for 40 min. The comet chip was then transferred to alkaline buffer for 40 min, with fresh alkaline buffer being replaced after 20 min. The comet chip was subjected to electrophoresis for 50 min with a constant setting of 22 V. Immediately after electrophoresis, the comet chip was neutralized in pH 7.5 400 mM Tris-HCl prior to staining with SYBR green diluted in 40 mM Tris-HCl. Comet chips were imaged using the BioTek Cytation7 cell imaging multimode reader, and tail DNA was measured using the Trevigen Comet Assay Analysis Software.
Measuring the Impact of Temperature on Cell Viability
To determine the role that temperature plays on cell viability, the temperature inside the MelodySusie (MS) unit was measured during the acute exposure protocol. Initially, the temperature was tested using a generic thermometer at two locations within the MS unit. From this, it was determined that indeed there were temperature changes. To measure temperature fluctuation more precisely, we used a pico-temperature measurement system. This apparatus measured temperature in the LUDIS and MS units over a 20-min irradiation in 8 distinct well locations. Temperatures were within the 40-65°C range. To determine the impact the high temperature had on cells, both keratinocyte and fibroblast cells were seeded in 96-well plates at 104 cells/well and were left overnight in a 37°C incubator. To replicate and examine the effect of the temperature changes in the MS device in the absence of UVA, cells were then placed in an incubator set to 55°C for 20 min (twice, with a 60-min break between treatments to replicate the acute exposure protocol), or 31°C for 15 min (to align with the 6 × 30 sec salon exposure schedule), before being returned to a 37°C incubator. Measurements were conducted 48 hr after final exposure; NHDF viability was measured using an XTT Cell Viability Assay Kit (Biotium, Fremont, CA, USA). Plates were incubated at 37°C for 4 hr, and absorbance was read at 450 nm using the BioTek Cytation7 cell imaging multimode reader. E6/E7 viability was measured using DAPI stain after cells had been fixed with 4% paraformaldehyde and permeated with 0.2% Triton. Plates were incubated for 5 min at 4°C, and cell count was taken using the BioTek Cytation7 cell imaging multimode reader. Finally, to determine the impact of temperature changes within the MS unit (localized regions of elevated temperature), a plate of cells was covered in aluminum foil and placed in the MS unit for an acute 20-min protocol. Cells were allowed to recover for 48 hr before cell viability was measured.
Statistical Analysis and Assay Controls
Statistical evaluation of data was conducted initially using Excel (Microsoft) to annotate and organize raw data that was generated from the cell viability and comet analysis assays. The data was then exported to a dedicated statistical analysis and visualization software (Prism version 10.1.2, Graphpad, Los Angeles, CA). Statistical approaches used in this research include one-way ANOVA with post-hoc analysis unless otherwise specified. In Figure 1(F), the modeling was changed to a mixed effect analysis (REML) without the assumption of sphericity and Dunnett’s multiple comparison testing and resulted in the same adjusted P value of <0.0001. Figures 4(D) and 4(G) show the two sets of data on each of the graphs that passed normality testing (Shapiro–Wilk, Pearson, Anderson–Darling, and Kolomogorov), with each irradiation time point compared between acute and chronic exposures using one way ANOVA with multiple comparisons corrected for using Tukey post-hoc testing. Statistical significance was measured in Figure 4(D), A9.8 vs. C9.8 adjusted P value of <.0001, A14.2 vs. C14.2 adjusted P value of .013 (*). In Figure 4(G), A9.8 vs. C9.8 adjusted P value of <.0001, A14.2 vs. C14.2 adjusted P value of .0047 (**). The Figure 5(F) data were analyzed using Kruskal–Wallis multiple comparison testing with Dunn’s post-hoc test. Statistical significance was gauged as * = P < .05, ** = P < .01, *** = P < .001, and **** = P < .0001.
The DNA damage measurements were conducted using company-established Standard Operating Procedures (SOPs), including the inclusion of positive and negative controls as previously described in Ref. 12 and also in Ref. 11. One of the many innovations presented herein is the use of stable DNA damage controls. These positive controls have been described in the publication 12 above and are cells that have been exposed to etoposide and then fixed. They have a stable level of DNA damage that is used to normalize multiple comet assays. There is no negative control for the comet assay as all cells have some level of endogenous DNA damage, nor is a blank no-cell control appropriate for this methodology.
Results
Measurement of Cell Viability in Human Cells after Exposure to UVA from a Commercial Nail Dryer Unit
We investigated the irradiation characteristics of the MelodySusie UV-curing unit, exposing non-isogenic human keratinocytes (E6/E7) and normal human dermal fibroblasts (NHDF) to three different treatment protocols (Figure 1). The acute treatment protocol was 2 × 20-min irradiation with 60 min between the two treatments, while chronic exposure protocol was 3 × 20-min exposures with 24-hr recovery between each of the three treatments.
9
The third treatment protocol was based on a typical salon setting, with each (or all) nail(s) cured immediately after the coating is applied (5 × 30 sec) and 2 min between treatments (during which time the next nail is coated). The last cure (30 sec, 6 × 30 sec total) hardens the protective sealant coating on all five nails; this salon protocol is depicted in Figure 1. The MelodySusie (MS) unit has a total light output of 54 W and 3 pre-programmed durations of continuous exposure (30, 60, and 90 sec). Depicted in Figure 2(A), the LED configuration of the machine is arch-shaped to sit above the locations of each nail of a palm-down flat hand. The curing LEDs emit UVA at 365 nm but are not all at a uniform height or density, resulting in the unit emitting approximately three times more UVA (5 to 15 mW/cm2/sec) at the rear than at the front (Figure 2(B)), as measured using a handheld photometer with a microscale UVA-calibrated detector. We irradiated human-derived keratinocytes, and cell viability was measured after 48 hr of recovery (Figure 2(C)-(F)). After acute exposure, high levels of cell death correlated with LED location in the MS unit (Figure 2(C)). That is, no loss of viability was measured at the front of the unit (wells H5-7). In contrast, viability dropped to below 10% of control values in wells directly below the main arch-shaped array of LEDs (R) in the unit. A similar pattern of cell viability was achieved when using the 3-day chronic exposure schedule (Figure 2(D)). While these exposure protocols are informative regarding the general cytotoxicity of nail dryer LEDs, they do not model a typical level of LED exposure likely encountered when using these units in a nail salon setting. To reproduce the conditions more likely to be encountered in a nail salon, we exposed keratinocytes to a third experimental paradigm (6 × 30 sec exposure with 120 sec between exposures). This lower level of irradiation did not cause a reduction in cell viability (Figure 2(E)-(F)). Further, the three exposure protocols resulted in significantly different viability results (Figure 2(F)). The chronic exposure protocol reduced average viability to the greatest extent (25 ± 9%; P < 0.0001) compared to acute (39 ± 16%) and salon (142 ± 11%) irradiation protocols. In brief, when the unit was used according to the manufacturer’s specifications, there was no reduction in cell viability. Human keratinocyte viability after UVA exposure. (A) The underside of the MelodySusie (MS) nail dryer. The LED configuration is based on the shape of the human hand. The black arrow indicates that the direction hand is inserted into the unit. F = front, R = rear. (B) The area at the rear (R) of the unit is exposed to approximately three times the UV intensity of the front (F) of the unit. (C) Cell viability measured 48 hr after acute exposure (defined in Figure 1). The greatest reduction in cell viability occurred in an arch-like shape consistent with the location and concentration of LEDs in the unit. (D) Chronic exposure produced a similar pattern of cell viability. (E) In contrast, no loss of cell viability was measured when using salon exposure. (F) Comparison of the average viability after the three exposure protocols. Each set consists of 192 points from the duplicate experiments depicted in (C), (D), and (E). **** = P < .0001 using a mixed effect analysis (REML) without the assumption of sphericity and Dunnett’s multiple comparison testing. Error bars represent mean ± SD.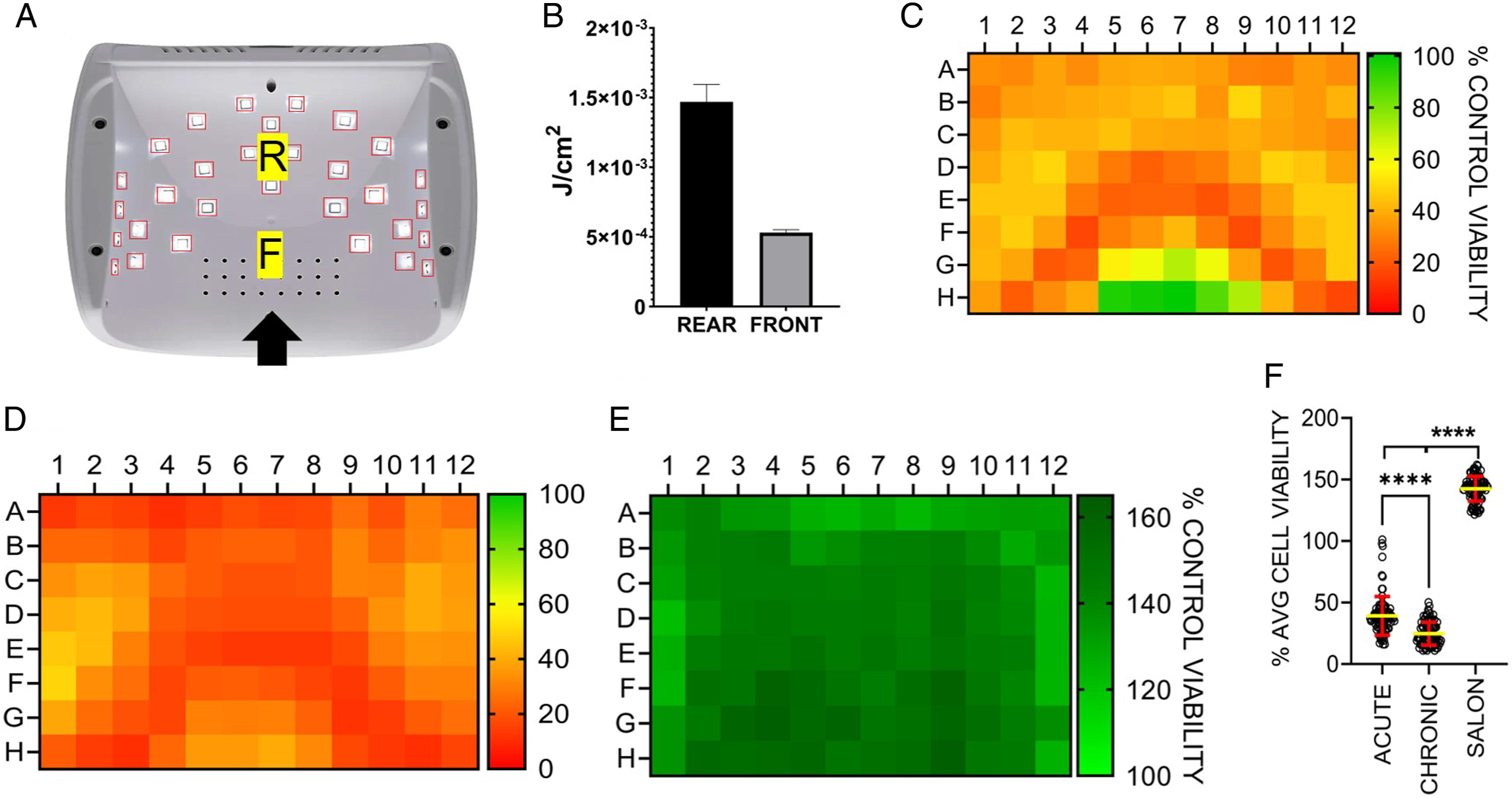
UVA can penetrate through the epidermis into the lower dermal layer containing predominantly fibroblasts. Using the previously described three treatment schedules (Figure 1), we investigated the impact of the UVA exposure on normal human dermal fibroblast (NHDF) cells. Acute exposure resulted in an approximately 50% decrease in fibroblast viability (Figure 3(A)) that, unlike keratinocytes, was independent of location of cell wells in the dryer. In contrast, chronic treatment of the NHDF cells resulted in a pattern of reduced viability in wells directly below the UVA LEDs (Figure 3(B)), as previously seen in the keratinocytes (Figure 2(D)). Under salon conditions, the NHDF cells showed a reduction in viability correlative to positional LED density (Figure 3(C)), although the pattern was less defined than that observed using the chronic treatment protocol (Figure 3(B); summarized in Table 1). Normal human dermal fibroblast exposure to UVA. Cell Viability After the Three Different Exposure Protocols.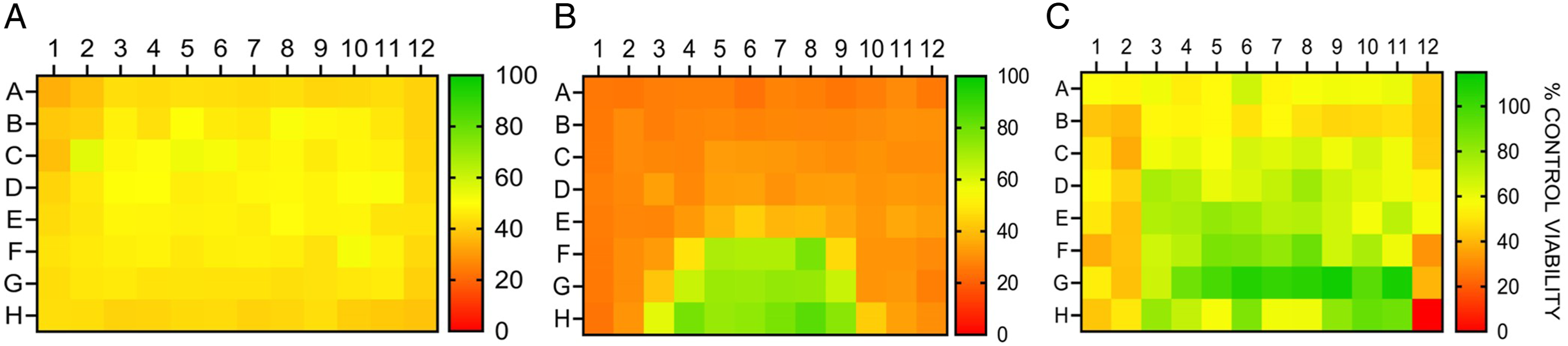

Determining the Amount of UVA Exposure at the Irradiation Surface of the MS Unit
Previous methods to accurately measure the output from nail dryers have yielded varied results.
13
This variability can be partly attributed to the non-uniform light distribution and density in commercial nail dryers (Figure 2(A)). We therefore evaluated different methods to measure the fluency inside the MS unit. Initially, we trialed moving a UV-detecting sensor in a designated grid pattern within the MS irradiation zone. However, the size of the sensor affected the light scatter patterns within the machine, and the sensor was not at the same irradiation height as the cells. We then sought to correlate the output to a device with a previously determined UVA output. The LED UV-DNA damage induction system (LUDIS) is an in-house patented prototype that was built by our consortium as part of a body of research investigating the impact of UVA on human skin. The LUDIS LEDs are in a 96-well configuration (Figure 4(A)). Similar to the MS unit, the LUDIS contains UVA (365 nm)-emitting LED bulbs. Each LUDIS LED has an individual light-directing channel (light cone) to prevent light from scattering into adjacent wells. We tested how the keratinocytes and fibroblasts responded to UVA irradiation from the LUDIS using the Acute exposure schedule. Cells were exposed to 0 (Rows A-B), 4.5 (C-D), 9.6 (E-F), or 14.2 (G-H) J/cm2 (Figure 4(A)). Keratinocyte viability after LUDIS UVA exposure was similar to the MS results (Figure 4(B)-(C)) with chronic treatment inducing greater loss of cell viability than acute treatment (Figure 4(D)) at 9.6 and 14.2 J/cm2. The E6/E7 used in this study is particularly amenable to high-throughput screening platforms but has aberrant p53 and pRB pathways.
14
The DNA damage induction profile of E6/E7 cells after UVA1 irradiation (acute) was compared to normal human adult keratinocytes (NHEKa) that were not immortalized (ATCC, PCS-200-011) (Figure S1). The amount of UVA1-induced DNA damage was higher in the NHEKa cells but with a comparable pattern of response. The LUDIS exposure experiments were also carried out using fibroblast cells under identical experimental conditions. As reported for the acute MS fibroblast results, cells did not respond to the acute UVA exposure in a linear manner with increasing fluency correlating with decreasing viability. Rather, the fibroblast gave a binary response, with the acute lowest exposure (4.5 J/cm2) and high exposure (14.2 J/cm2) inducing a comparable reduction in overall cell viability (Figure 4(E)). In contrast, the chronic LUDIS exposure in the fibroblasts did induce a reduction in cell viability correlating with UVA fluency (Figures 4(F) and 4(G)). The LUDIS linear response data from the keratinocytes was then used to create standard curves (Figure S2A and S2B). The linear regression equations derived from these curves were then used to calculate the absolute UVA exposure level (J/cm2) for each well in the MS unit (Figure S3A and S3B). However, depicted as in Figure S3C, the two exposure protocols significantly differed (overall 2.6 ± 4.0 J/cm2), particularly along the left edge of the plate (columns 1 and 2, difference 4.4 ± 1.5 J/cm2) and in the UV-LED arch area (5.4 ± 3.3 J/cm2). From these data, we concluded that a second endpoint would be needed to enable cross-comparison, potentially improving the precision of the predictive approach. Using a HTP UVA irradiation device: 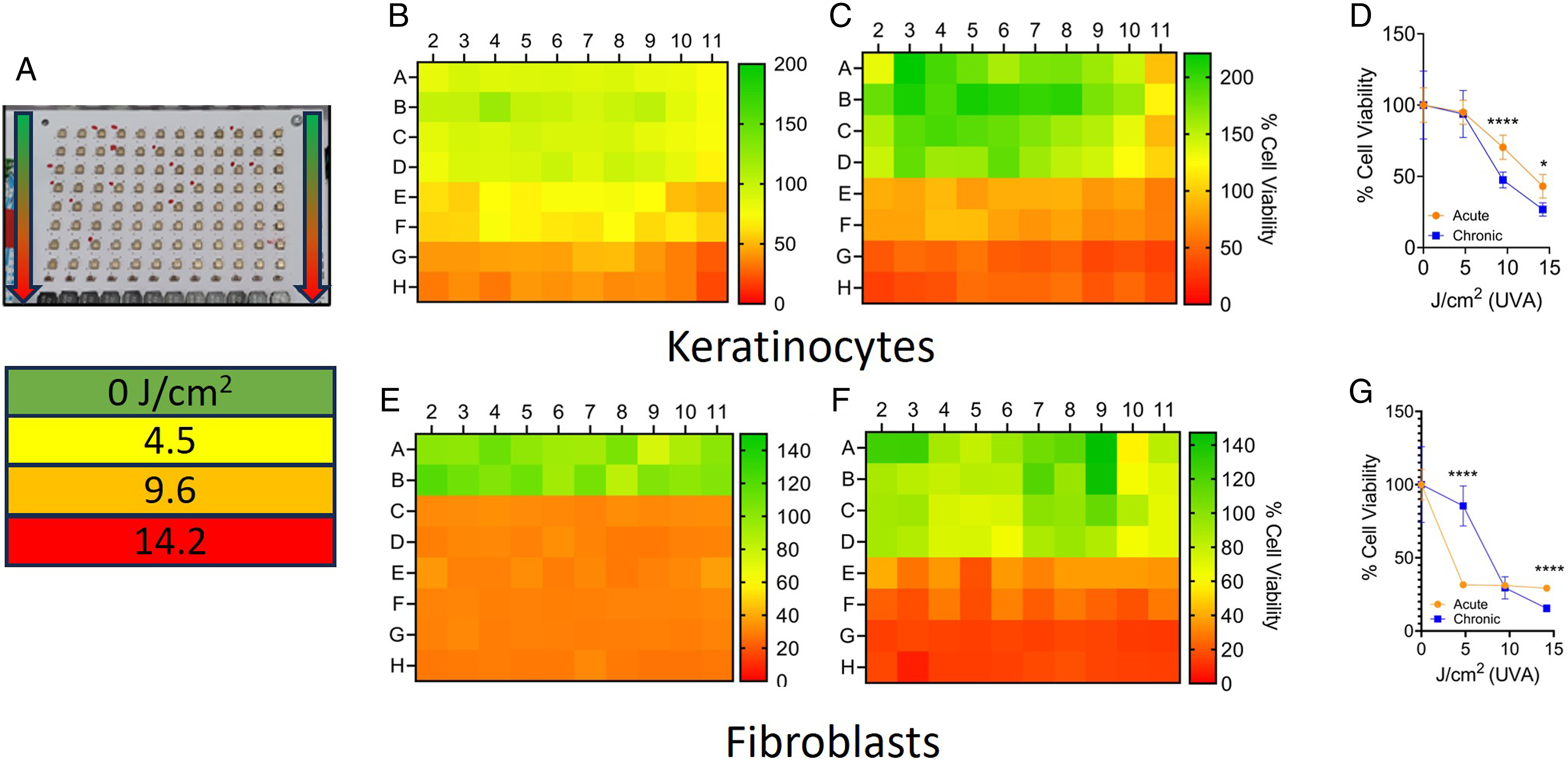
The Role of Oxidative DNA Damage in the Loss of Viability after UVA Exposure
Oxidative DNA damage was evaluated as a second endpoint to correlate UVA exposure between the LUDIS and MS units. UVA can cause DNA damage through indirect mechanisms, including the induction of reactive oxygen species (ROS). Intracellular superoxide, a potent ROS molecule, was measured after exposure to UVA (Figure 5(A)). As per previous reports,
9
UVA exposure elevated levels of superoxide. This impact was observed in both fibroblasts and keratinocytes, with levels of induced superoxide plateauing at higher fluency (>5 J/cm2). We investigated whether levels of DNA damage correlated with superoxide induction using single cell gel electrophoresis (SCGE, aka comet assay). SCGE allows for the visualization of DNA damage, including lesions caused by oxidative damage, such as AP-sites and single-stranded breaks (SSB). Acute exposure of keratinocytes using LUDIS resulted in significantly lower levels (P = .043) of DNA damage than that of 3-day chronic treatment (Figure 5(B)). We then replicated the experiment in the MS unit. The pattern of DNA damage was correlative with the position of the LEDs; however, there were regions of high DNA damage at the periphery of the plate after any of the 3 (acute, chronic, and salon) irradiation protocols (Figure 5(C)–5(E)). Overall, the salon exposure schedule caused significantly less DNA damage than that of the acute (P > .001) and 3-day chronic (P > .0001) treatments. Of note, the chronic exposure caused more DNA damage than measured after the acute treatment (Figure 5(F)). In comparison to the keratinocytes, the LUDIS induced significantly (P > .0001) higher levels of DNA damage in the fibroblasts after acute and chronic exposure (Figure 5(G)). Using the MS for fibroblast, UVA irradiation caused many wells to reach DNA damage levels, which were deemed to be beyond assay linearity (>60%) (Figure 5(H)–(J), (K)). Because the acute and chronic measurements went beyond assay linearity, further statistical analysis of the fibroblast data was not appropriate. Overall, the fibroblasts were more susceptible to UVA-induced DNA damage compared to the keratinocytes (P > .0001), yet the pattern of DNA damage induction did not clearly correlate with the arch-shape of the LEDs. We used the acute LUDIS keratinocyte data to determine whether the measured linear DNA damage (r2 = 0.98) could correlate and predict the UVA output of the MS unit. To this end, a standard curve was created from the LUDIS DNA damage results (not shown), and this curve was used to predict the UVA exposure in the MS unit (Figure S4A). The DNA damage levels did not correlate with the position of the LEDs; rather high levels of DNA damage were measured at the plate peripherals. Hence, the fluency calculations also estimate high UVA levels in these areas of the plate. This irradiation prediction was compared to the acute keratinocyte cell viability result (Figure S4B) with the difference between the two results presented as Figure S4C. As with the cell viability correlation, but using DNA damage as an endpoint, we were not able to predict the level of UVA exposure within an acceptable level of accuracy. Specifically, the calculations overestimated the amount of DNA damage around the periphery of the plate, in particular in row B of the acute treatment; this row sits closest to the back of the MS unit. As with the cell viability predictions, the DNA damage prediction data suggested unknown additional factors beyond UVA exposure were influencing both DNA damage and cell viability in the MS unit. Impact of UVA exposure on associated metrics. 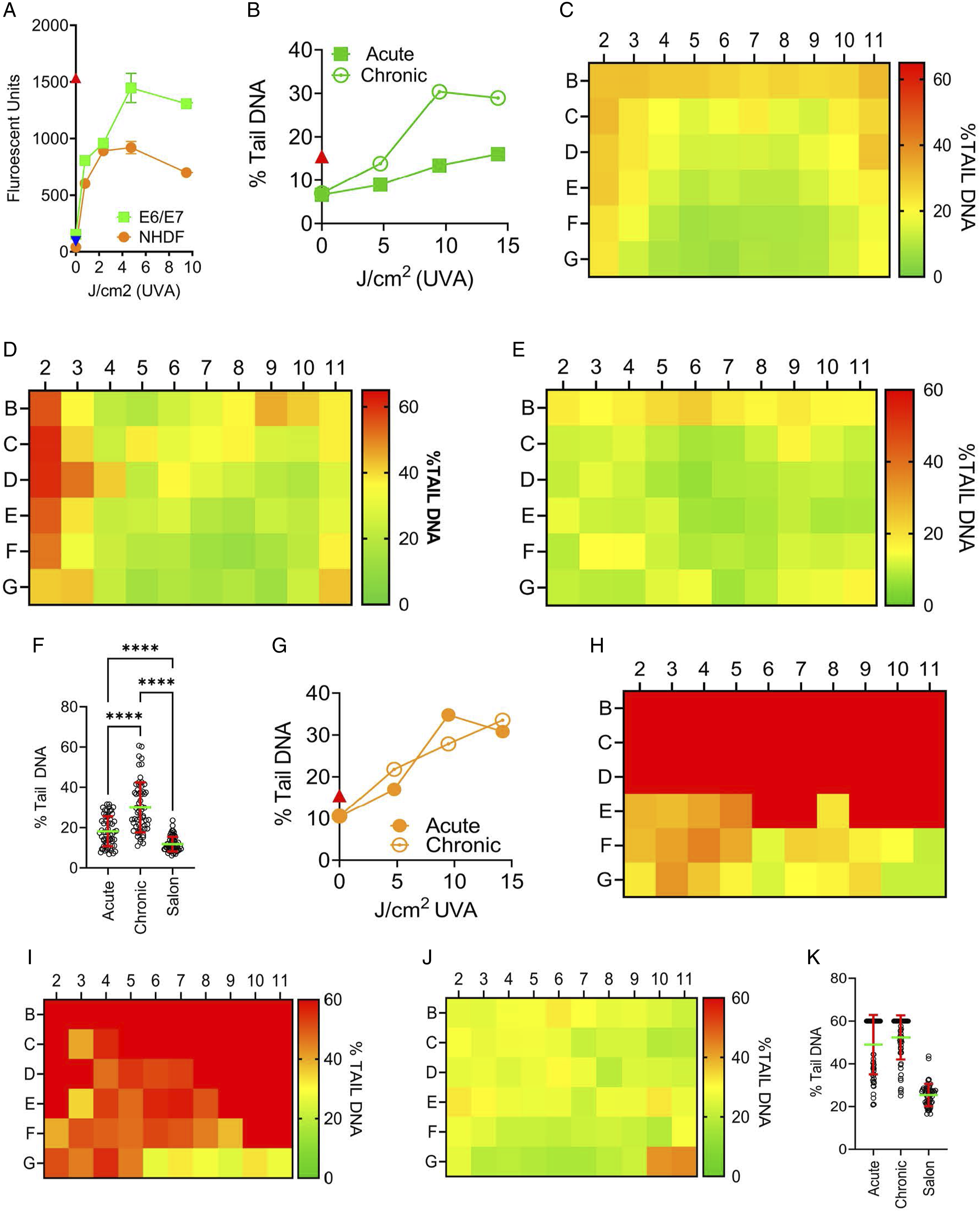
Considering the lack of onboard cooling in the MS, we investigated whether the zone of irradiation inside the unit had fluctuations in temperature. To measure changes in temperature, we used a thermometer inside the MS unit. Towards the opening of the machine (F), temperatures increased over the 20-min exposure duration to a maximum recorded heat of 45°C (Figure 6(A)). This correlated to the area with the highest viability after exposure (Figures 2 and 3). When the probe was placed at the back of the unit, the temperature reached a maximum of 55°C (Figure 6(A)). The magnitude of temperature change was unanticipated and required more precise analysis. To this end, a digital microprobe temperature detection system was used. The internal well temperature rose above 60°C during the acute treatment protocol in the MS (Figure 6(B)). To determine the impact this temperature change may have had on the cell viability, a plate of fibroblast cells was put in an incubator pre-warmed to 55°C for 20 min. The fibroblasts showed a well, row, and column-independent reduction in viability with an average reduction in viability of approximately 50% (Figure S5A). The incubator cell viability experiment was repeated using the salon protocol. This also reduced the cell viability by an average of 25% (Figure S5B). In contrast to the position-independent decrease in cell viability observed in the fibroblasts, the keratinocytes showed an exposure pattern that showed the greatest loss of viability (near 100% loss) at the peripherals of the plate while the center of the plate had a 50% loss of viability (Figure S5C); in these cells, there was a clear gradient from the outside to the center of the plate, presumably as a result of more rapid warming of peripheral wells in the incubator. To complete the set of temperature experiments, we covered a plate of fibroblast cells in a single layer of aluminum foil and exposed the plate in the MS under the acute protocol conditions. Aluminum foil was selected because it transfers heat readily and completely blocks the UV light. After exposure, the cell recovered for 48 hr, and cell viability was measured (Figure 6(D)). A loss of cell viability was measured at the peripherals of the plate similar to Figure S5C, with the greatest loss of cell viability measured at the left peripheral side of the plate. The experiment confirmed that using the MS induces an internal temperature increase that impacts cell viability independently of UVA exposure. This temperature increase is not ubiquitous throughout the unit and impacts both UVA-induced loss of cell viability and also, to a greater extent, levels of induced DNA damage. In the research conducted by Zhivagui et al.,
9
the authors used a larger format 6-well plate rather than the 96-well format used herein. To determine whether the temperature fluctuations were an artefact of using the smaller well format, we also measured the temperature using a generic 6-well plate (Figure S6A-B). The wells irradiated at the front of the unit (A1, A2, and A3) all had a significant increase in temperature after 20 min in the nail dryer. This was highest for well A3 that had a peak temperature of over 50°C. In contrast, the three rear wells did not have a significant increase, suggesting that differences in heat dissipation patterns and the pattern of LED lights in the unit may play a major role in the elevated temperatures measured in both plate formats. The ramifications of elevated temperature on 2D mammalian cell culture and DNA damage are well established and will be discussed further herein. The influence of heat fluctuation on cell viability. 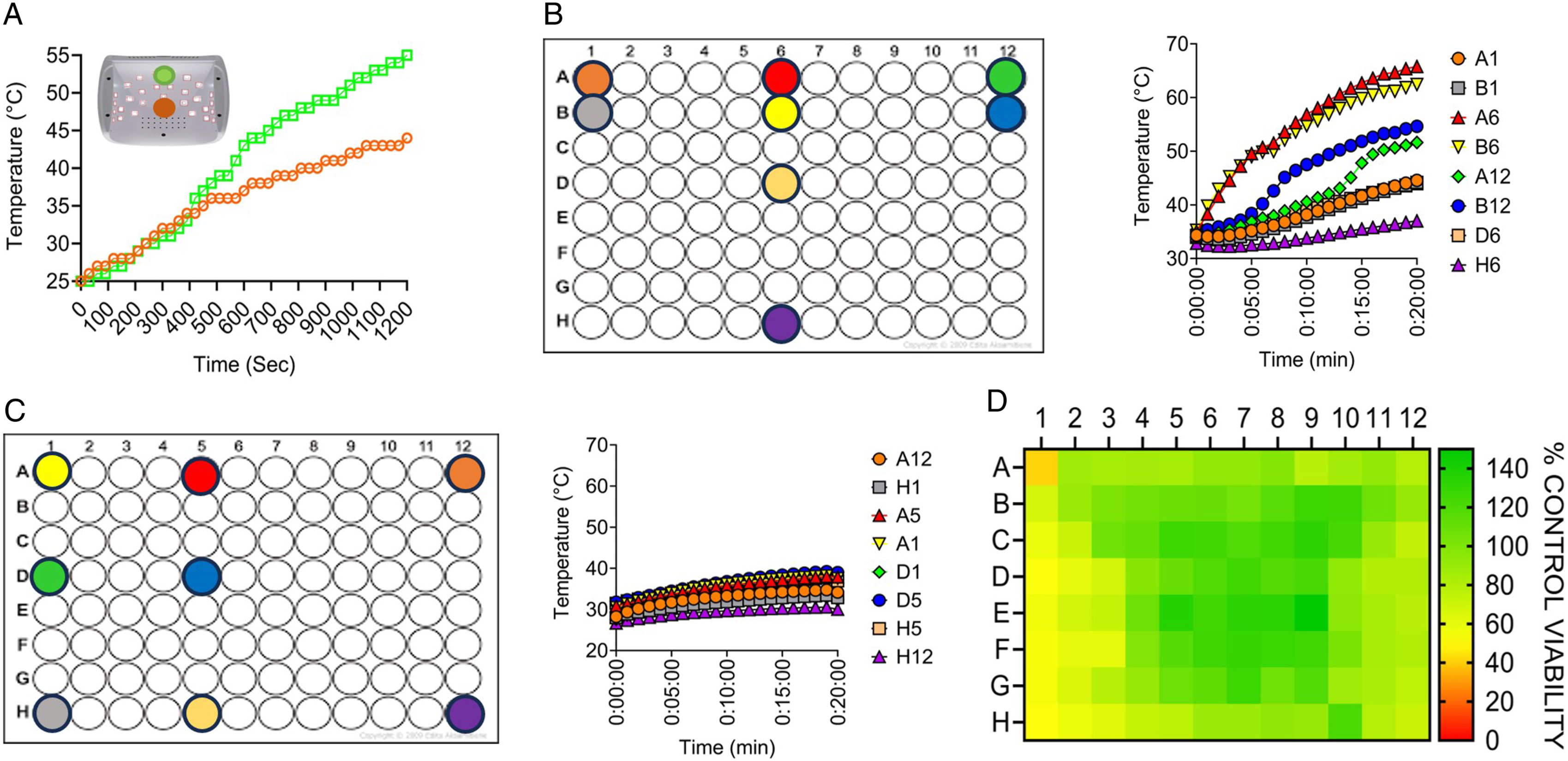
Discussion
The primary objective of this research was to develop a better approach to measuring the amount of UV radiation in commercial nail dryer units. A secondary objective of the research was to determine whether using the unit under realistic salon conditions would induce changes in biological endpoints associated with cell cytotoxicity and DNA damage. The exposure protocols were determined using previous research and suggested manufacturer’s specifications. We used human skin-derived 2D cell lines to measure UVA exposure patterns and biological endpoints. The two cell types investigated responded very differently with acute exposure, inducing a decrease in fibroblast cell viability with no correlation to the level of UVA fluency. The same outcome was reported using the LUDIS platform with no correlation between acute cell viability and UV fluency in the fibroblasts. In contrast, after chronic exposure, a clearly defined LED-radiation arch configuration was observed using fibroblasts. The arch pattern was also observed after the salon treatment, suggesting that the pan decrease seen in the fibroblast acute treatment is a product of the treatment regime, specifically the two back-to-back irradiation treatments within a short period of time. We were not able to delineate what impact that elevated temperature in the MS unit played in the loss of cell viability. We do report an analogous decrease in fibroblast cell viability using acute heat treatment alone. While this may suggest that temperature played a significant role in the UVA-induced loss of cell viability in these experiments, it does not explain why the same pan decrease was also observed using the LUDIS. The LUDIS is an enclosed unit specifically designed to ensure samples do not increase in temperature during irradiation. This is achieved with air cooling below and above the plate while irradiating, and the LUDIS has been previously tested for well-temperature fluctuation. Further, the acute unirradiated wells in the LUDIS measured no decrease in cell viability despite being adjacent to wells that had large decreases in cell viability. This result suggests that ineffective DNA repair between treatments may cause the cells to spiral into apoptosis, an impact not seen in the keratinocytes. The E6/E7 cells used in the study have aberrant p53 and pRB pathways, which have consequences for the downstream impact of any induced DNA damage, particularly the cellular decision-making regarding DNA repair and the induction of apoptosis. This in turn may affect the rate of skin cancer development. However, this study was largely focused on the induction of DNA damage rather than the downstream consequences of the DNA damage.
One of the main objectives of the presented research was to create a standard curve for cell viability after UVA exposure and use this curve to determine approximate UVA exposure for each well (region) during the MS irradiation schedule. In the fibroblasts, the non-linear decrease in viability precluded these cells from further analysis. In contrast, the LUDIS-exposed keratinocyte data had a strong linear regression correlating UV exposure and cell viability, yet this correlation could not be used to accurately predict UVA exposure levels in the MS unit. Specifically, the UVA exposure values for cell survival for LUDIS acute exposure provided a fluency value for the MS. However, the value was different when comparing the LUDIS and MS for the 3-day chronic exposure schedule. This was also the case when using DNA damage as an endpoint for comparing the LUDIS and MS; again, using linear regression provided starkly different estimates of equivalent UVA fluency/dose for acute vs. chronic exposure. It appeared that the temperature differential in the MS plate was a major additional variable that altered cell viability and genotoxicity of cells exposed to the MS unit. The evidence here suggests that elevated temperature analogous to hyperthermic conditions contributed to the induction of cell death and DNA damage in the MS unit.
With respect to the role of temperature on cytotoxicity and genotoxicity, it is well established that hyperthermia influences DNA repair processes. Hyperthermia has been applied in-clinic for decades to sensitize cancer cells to chemotherapeutics that induce DNA damage. 15 The molecular mechanism behind the sensitization is multifaceted and may include the direct induction of DNA damage but also the concurrent inhibition of DNA repair. While the pathways of heat-induced direct induction of DNA damage remain largely to be determined, Warters et al. showed that the apurinic (AP) site could be induced at temperatures between 43 and 48°C.16,17 This corroborated earlier work showing that there were elevated levels of DNA strand breaks in HeLa cells exposed to temperatures between 43 and 45°C. 18 Despite these early sporadic reports, it is more probable that higher levels of DNA damage after hyperthermia are not caused by direct induction but rather by the inhibition of cellular processes at elevated temperatures. These include the deactivation or inhibition of multiple, if not all, repair pathways. In previous research, it has been determined that nail dryer-induced UVA exposure using a 2D cell culture model resulted in high levels of C>A mutations corresponding to COSMIC single-base substitution signatures SBS18 and SBS36, as described in Ref. 9. Both signatures have been attributed to exposure to elevated levels of ROS. The main repair pathway for small non-helix-distorting oxidative lesions is base excision repair (BER). The BER pathway has been reported to be inhibited at temperatures considerably lower than those reported herein using the MS unit (55°C). C>A nucleotide substitutions are associated with the oxidative lesion 8-oxo-dG processed by 8-oxoguanine DNA glycosylase (OGG1). Cellular glycosylase activity was reported to be affected by hyperthermic temperature (42°C), inactivating OGG1 by triggering proteasomal-mediated degradation and exit out of the nucleus.17,19 The cellular effect of the hypothermic-mediated OGG1 loss was the sensitization of the cell line to DNA-damaging agents. Hyperthermia may also preferentially affect the core BER protein DNA polymerase β (Polβ). The mechanism of this inhibition may be related to the influence of BER scaffold protein XRCC1 on the overall BER complex. Unbound XRCC1 interacts with the chaperone protein heat shock protein 90 (HSP90). Further, the dynamic interaction between Polβ- XRCC1- HSP90 regulates the BER subpathway choice. 20 This is supported by studies using a variety of DNA-damaging agents, all showing that Polβ is inhibited at temperatures as low as 41°C.21-23 DNA repair was not measured in our studies; however, we did measure elevated DNA damage levels in the areas of the plate that have the highest temperature. These regions of elevated damage did not correlate with the highest levels of UVA irradiation as expected. The finding that heat plays a role in the DNA damage profile induced by the MS limits any physiological relevance that can be derived from the research.
The skin has evolved mechanisms to deal with heat and maintain homeostasis in elevated temperatures, including vasodilation and sweating. In an example, such as using a nail dryer, the body can increase the blood flow rate to quickly dissipate heat from the area to maintain stable tissue temperature. Blood flow to the skin can range from nearly zero in extreme cold to 6-7 L/min during extreme heat stress. 24 In contrast, 2D in vitro cellular systems are exquisitely sensitive to temperature fluctuations and possess only a limited strategy for heat dissipation. We determined that using 2D cell culture models in nail dryers and extrapolating the result to human health metrics had serious flaws that had not been previously addressed. The discovery of the elevated temperature in the MS unit, a factor not equivalent throughout the apparatus, prevented the accurate calculation of UVA fluency in the MS unit by correlation with a known UVA source.
The secondary objective of this research was to compare different UVA exposure protocols and investigate the potential biological impact of the MS unit. Many of the previous reports investigating UV nail dryers do not justify how UVA exposure parameters were calculated. For example, using the MS to irradiate the keratinocytes using chronic exposure induced greater than 70% cell death. This is similar to viability levels previously reported after chronic exposure. 9 However, it remains unclear why a nail dryer would be used for 20 min a day over three consecutive days. Similarly, the acute protocol (used herein and previously 9 ) consisted of 2 × 20-min treatments with 60 min between treatments; again, this is inconsistent with the normal use of a nail dryer. The downstream impact of these unconventional treatment protocols is the induction of high levels of cell death. There have been no reports of UV nail dyers used in salon conditions causing the user pain, redness, or swelling consistent with widespread cell death in the epidermal or dermal skin layers. As shown herein, using the machine according to the manufacturer’s specifications did not cause a loss of keratinocyte cell viability compared to controls. Questions remain regarding the human health impact that nail dryers may have on the user and nail technician. In response to the media interest regarding the Zhivagui finding, many salons are using protective measures including the use of UV-blocking eye protection for the technicians. Customers are now also being offered gloves with the top of the fingers cut off to protect the hand during drying protocols. It is still to be determined whether these mitigation strategies have any impact on any potential long-term UV skin damage caused by the units.
Supplemental Material
Supplemental Material - Temperature is a Key Factor Governing the Toxic Impact of UVR-Emitting Nail Dryers When Used on Human Skin Cells
Supplemental Material for Temperature is a Key Factor Governing the Toxic Impact of UVR-Emitting Nail Dryers When Used on Human Skin Cells by Elijah Finn, Lucia Dussan, Scott Rosenthal, Cynthia Simbulan-Rosenthal, Dean Rosenthal, and Peter Sykora in International Journal of Toxicology
Footnotes
Acknowledgements
Dr Peter Sykora and Dr Dean Rosenthal are principal investigators in the forementioned grant. ChatGPT was used to originally develop the framework for the introduction; however, this has been significantly edited, and very few of the original AI-derived sentence structures still remain in the document.
Author Contributions
EF, LD, and SR contributed to acquisition, analysis, and interpretation of data and gave final approval. CS-R and DR contributed to conception and design, critically revised the manuscript, and gave final approval. PS contributed to conception and design, contributed to analysis and interpretation of data, drafted the manuscript, critically revised the manuscript, and gave final approval.
Declaration of Conflicting interest
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr Peter Sykora is the CSO and director of Amelia Technologies LLC. None of the entities involved in this research have any commercial interest now or in the future regarding UV-emitting nail devices.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The development of the LUDIS was supported in part by grant R42ES032435 from the NIEHS.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.