
Abstract
Etripamil is a calcium channel blocker currently in Phase 3 trials for the treatment of paroxysmal supraventricular tachycardia (PSVT). Systemic and local toxicity following once-weekly intranasal administration of etripamil was evaluated in cynomolgus macaques to support clinical development. Groups of animals (N = 8, 4 males and 4 females) were administered etripamil into the left nostril weekly at dose levels of 0 (vehicle), 1.9, 3.8, or 5.7 mg/kg/dose for 26 doses. Persistence, reversibility, and progression of findings were examined following a 28-day recovery period. Clinical signs were transient and were related to the intranasal administration (e.g., nasal discharge, sneezing, etc.) of etripamil. There were no macroscopic or systemic microscopic findings at any dose. Etripamil-related adaptive and reactive local changes affecting the nasal cavity, larynx, and nasopharynx were observed at ≥1.9 mg/kg/dose. Minimal to severe dose-dependent nasal epithelial damage was observed, mainly affecting respiratory and transitional epithelium. Following the 28-day recovery period, microscopic changes were confined to the left nasal cavity and nasopharynx. These changes were significantly lower in incidence and severity, with noticeable reversal of the adaptive and reactive changes, indicating partial to complete recovery of the epithelial lining. Based on the lack of systemic toxicity and the minimal and transient nasal changes, the systemic, no observable adverse effect level (NOAEL) of etripamil in monkeys was the high dose, 5.7 mg/kg/dose. The NOAEL for local toxicity was 1.9 mg/kg/dose. Collectively, these data support further study of etripamil in human trials as a potential treatment for PSVT.
Keywords
Introduction
Paroxysmal supraventricular tachycardia (PSVT) defines a subset of tachycardias that are intermittent, sustained (>30 seconds), and have a regular ventricular response.1,2 From 2010 to 2015, the prevalence of PSVT in the United States was estimated to be 168 per 100,000 population. 3 Atrioventricular nodal re-entrant tachycardia (AVNRT) and atrioventricular reciprocating tachycardia (AVRT) are often the causes of PSVT. The re-entrant circuit may develop into one that is repeatedly activated, yielding a prolonged reentrant tachycardia. While PSVT is typically not life-threatening, patients often experience poor quality of life due to symptoms that may include chest pain, palpitations, lightheadedness, and anxiety. The factors associated with PSVT include old age, being female, and a history of cardiovascular disorders. 3
Vagal maneuvers are the first-line treatment for PSVT, as they slow down the sinoatrial node (SA) activity and AV nodal conduction by stimulating baroreceptors in the aorta, thereby triggering increased vagal activity. 4 However, when self-performed, these maneuvers rarely terminate PSVT. 4 An effective treatment to terminate PSVT involves the use of intravenous (IV) adenosine, which activates K channels and reduces calcium influx by antagonizing adenyl cyclase, resulting in slowing of sinus rate and increased AV conduction delay. 5 Side effects of IV adenosine include palpitations, chest pain, and light headedness. Calcium channel blockers (CCBs) can also be administered intravenously to terminate PSVT 5 ; however, IV CCBs may be either ineffective or intolerable due to hypotension, and they also require medical supervision in a hospital setting. 5 An unmet treatment need in PSVT is the development of a fast-acting drug that could be self-administered by patients outside of the healthcare setting and without access to medical supervision.
Etripamil is a dihydropyridine calcium channel blocker (CCB) that is fast-acting with a short half-life of 2.5 hours 6 and is metabolized by serum esterases into MSP-2030, an inactive carboxylic acid metabolite. 6 The presence of an ester moiety on etripamil allows for rapid metabolism by serum esterases, decreasing the half-life and decreasing the time during which side effects might be present.7,8 Etripamil is currently in clinical development as a self-administered intranasal treatment and is conveniently utilized outside of the healthcare setting.
Moreover, etripamil has demonstrated rapid absorption by nasal mucosa in Phase 1 studies 9 and has demonstrated success in the NODE1 clinical phase 2 trial for PSVT. In a randomized, double-blind, multicenter, placebo-controlled, dose-ranging study, this drug provided strong evidence of efficacy. 8 These results prompted the initiation of the NODE-301 Phase 3 trial to examine the efficacy and safety of etripamil in a 2-part investigation. Part 1 was a multi-center, double-blind, placebo-controlled study evaluating the effectiveness of 70 mg etripamil versus placebo. Part 2 (RAPID) enrolled new participants (n = 658) in addition to a cohort of participants who were carried forward from Part 1 (n = 34); these 34 participants did not have an episode of PSVT in Part 1 by the cut-off date (January 15, 2020) and therefore were unable to use etripamil. Two test doses of etripamil (70 mg × 2) were administered 10 minutes apart in all potential Part 2 participants during sinus rhythm to ensure tolerability. Next, participants were randomized 1:1 to receive either etripamil or placebo. If participants suspected symptoms of a PSVT episode, they initiated an ECG monitor that recorded for 5 hours and performed a vagal maneuver; if symptoms persisted, participants self-administered the study drug. In Part 2, participants had the option to self-administer a second dose of the study drug 10 minutes after the first dose due to continued PSVT symptoms. PSVT was converted to sinus rhythm by 30 minutes in 64% (63/99) of participants in the etripamil-treated group versus 31% (26/85) in the placebo-treated group (hazard ratio 2.62; 95% CI 1.66–4.15; P < 0.0001). This study demonstrated that etripamil was safe and well tolerated and supported continued development.
Additional studies are needed to characterize the toxicokinetic profile for etripamil in males and females. Thus, it is necessary to identify a closely related and suitable animal model for a better understanding of etripamil safety profile in humans. The purpose of the present study was to determine the potential local and systemic toxicity and toxicokinetic profile of etripamil, and its primary metabolite, MSP-2030, following once weekly intranasal administration to cynomolgus monkeys for 26 doses and to assess the reversibility of any changes following a 28-day recovery period.
Materials and Methods
Experimental Design.

Conc, concentration; M, male; F, female.
aMain phase animals were terminated on Day 177.
bRecovery phase animals were terminated on day 205.
cGroup 1 animals received the reference item/vehicle, 5 mM EDTA, in sterile water for injection, USP, adjusted to pH 4.4 ± 0.2.
Forty-four (44) cynomolgus monkeys (20 males and 20 females, and 2 spares per sex) of Chinese origin were used in this study. At the onset of dosing, the age of the animals ranged from 2.5 to 4.0 years. The body weights ranged from 2.5 to 3.1 kg and from 2.3 to 2.9 kg for males and females, respectively. Animals were group-housed (2/cage) in stainless steel monkey cages equipped with an automatic watering system but were separated during designated activities (including initial arrival and during the period of staggered dosing initiation). Animals were single-housed, when necessary (e.g., aggressiveness, poor health, etc.).
Following group assignment, all cages were clearly labeled with a color-coded cage card indicating at least the study number and animal number. Animals were single housed for at least 24 hours following each treatment. The animal room environment was controlled (targeted ranges: temperature 21 ± 3°C; relative humidity 50 ± 20%; 12 hours light, 12 hours dark, except during study plan-designated procedures). Temperature and relative humidity were monitored continuously. A standard certified commercial chow (Envigo Teklad Certified Hi-Fiber Primate Diet #7195C) was provided to the animals twice daily, except during designated procedures. Treats or fruits/vegetables were provided to the animals as part of the facility enrichment program. As the animals were already acclimated to the laboratory environment, at least 13 days were allowed between transfer of the animals and the start of treatment. Animals were acclimated to the administration procedure, using 100 μL of saline in the BD Accuspray delivery device once daily, on the 3 days prior to the start of the treatment period. During the acclimation period, males and females were separately assigned to dose groups by a randomizing stratification system based on body weight. For logistical reasons, the animals were assigned to replicate subgroups, for which dosing was performed on consecutive days. Cage-side clinical signs (ill health, behavioral changes, etc.) were recorded twice daily throughout the study.
A detailed clinical examination was performed on each animal prior to animal assignment, then weekly starting on Day 8, and on the day of necropsy. The nasal cavity (both nostrils) was examined for all animals (main phase and recovery phase), using an otoscope, prior to the acclimation to the administration procedure, Day 1, Day 8 and on Weeks 4, 5, 6, 8, 12, 16, 20 and 25, approximately 1-hour post-dose. In addition, the nasal cavity (both nostrils) was examined from recovery phase animals near the end of the recovery period (Week 30). Of note, priority was given to the blood collection at 60 minutes after dosing over the examination of the nasal cavity, which was performed after the blood collection.
The study conduct complied with the OECD Principles of Good Laboratory Practice (ENV/MC/CHEM(98) 17) as accepted by Regulatory Authorities throughout the European Community, United States of America (FDA) and Japan (MHL W). The facilities that conducted the studies were in compliance with national or regional animal welfare regulatory authorities and are AAALAC-accredited. Local research animal ethics committees reviewed and approved the work with animals.
Clinical Pathology, Toxicokinetics, and Terminal Procedures
Clinical pathology evaluations (hematology, clinical chemistry, coagulation, and urinalysis) were performed on all animals once during the pre-dose period, on Week 4, Week 13, once towards the end of the dosing period (Week 25), and on recovery phase animals once towards the end of the recovery period (Week 29). During the main phase, the urine was collected overnight following the dosing and the blood collection was performed at least 24 hours post-dose. Blood samples were collected from the femoral vein and urine was collected overnight. Blood samples were collected from all animals at the following targeted time points relative to the Day 1, Day 92 and Day 176 dose administrations: pre-dose, at 1.5, 3.5, 5, 10, 30, 60 minutes, 4 hours, and 12 hours after dosing. Each blood sample was collected from the femoral vein into pre-chilled tubes containing sodium fluoride/potassium oxalate as anticoagulant and kept on wet ice pending centrifugation. Samples were centrifuged under refrigeration for 10 minutes within 30 minutes of blood collection. The resultant plasma was transferred into polypropylene tubes containing 10 μL of 0.5 N hydrochloric acid for each 200 μL of plasma, vortexed and then stored frozen pending shipment to Syneos Health (Quebec) for analysis and determination of toxicokinetic parameters. Plasma samples were analyzed using LC-MS/MS (API 5000), with ACE3 column (C18, 30 × 4.6 mm, 3 μm). Toxicokinetic parameters including terminal half-life (t1/2), time (tmax) to reach the maximum concentration (Cmax) after dosing were assessed by Phoenix pharmacokinetic software (Certara) using a noncompartmental approach consistent with intranasal administration.
For safety assessments, all animals were euthanized, and histopathology was performed at the scheduled termination, which occurred 24 hours after the last dose on Day 176. In these animals, mortality, clinical signs, changes in body weight, ophthalmology, electrocardiogram, clinical pathology (hematology, clinical chemistry, coagulation, and urinalysis), organ weights and macroscopic observations recorded. Tissues intended for histological examination were prepared by fixing the tissue in an appropriate buffered fixative, embedding in paraffin wax, sectioning and staining with hematoxylin and eosin. Histopathological examination was performed by the study pathologist, on the tissues from all main phase and recovery phase, and any gross abnormalities from animals of all groups. In addition, histological examination was performed on nasal cavities (4 sections/sinuses), larynx (3 levels), and pharynx for all study groups (1–4).
Data Analysis and Statistics
For each data set with more than two groups, a one-way analysis of variance (ANOVA) was performed, and the normality (Shapiro-Wilk test) and homogeneity (Levene test) were assessed. For the group mean comparisons between the reference item/vehicle treated group and etripamil treated groups, the Dunnett test was used to performed if the group variances were non-significant (P > 0.05).
Otherwise, if the group variances were determined to be heterogeneous (P ≤ 0.05), pairwise comparisons between the reference and etripamil treated groups were performed using a non-parametric Kruskal-Wallis test and Dunn test. For datasets with only two groups to compare (including the reference item/vehicle) and if the group variances were determined to be heterogeneous, a two-sample t test (5% level of significance) and Wilcoxon rank-sum test were used.
Each group comparison of the reference item/vehicle with an etripamil treated group was conducted via a two-sided test at the 5% significance level and the significant results were reported as either P ≤ 0.001, P ≤ 0.01, or P ≤ 0.05.
Results
No mortality occurred at any dose, in addition to no macroscopic or systemic microscopic abnormalities (including histology of the nasal cavity, larynx, and pharynx). Clinical signs at doses ≥1.9 mg/kg/dose included nasal discharge, sneezing, salivation, and vomiting, all of which were transient and related to the intranasal route of administration. Histological changes were observed in the nasal cavity, larynx, and pharynx of all animals treated with ≥1.9 mg/kg/dose but were not considered indicative of systemic toxicity (Figure 1). Changes in the nasal epithelium were minimal to severe, mainly affecting respiratory and transitional epithelium, and showed a dose-dependent trend toward increasing severity. Etripamil-related microscopic changes were confined to the left (administration site) nasal cavity and nasopharynx and were significantly lower in severity following the recovery period, suggesting ongoing recovery and a reversal of microscopic changes.
The systemic no observable adverse effect level (NOAEL) was determined to be the high dose of 5.7 mg/kg/dose based on the lack of systemic toxicity and transient, reversible nasal clinical observations. The NOAEL for local toxicity was determined to be the 1.9 mg/kg/dose. Microscopic evaluation of tissue samples following drug administration and 28-day recovery period. (A) shows normal respiratory epithelium overlying the nasal septum with numerous goblet cells. (B) At the end of the 6-month treatment period, etripamil-related microscopic observations were confined to the left nasal cavity (administration site), nasopharynx, and larynx. Histologically, in the nasal cavity of treated animals and when considering all four treatments as a whole, there was a dose-dependent increasing severity of erosion/ulceration of respiratory, transitional, and olfactory epithelium, as well as those associated with chronic inflammation. Also, a mild to moderate area of chronic inflammation of the respiratory epithelium of the nasal septum is shown (asterisk) with attenuation of the epithelium and erosion (arrow). (C) Following the 28 day, etripamil-related microscopic observations were confined to the left nasal cavity and nasopharynx. In general, the respiratory, transitional, olfactory epithelium occasionally exhibited the changes noted at the terminal euthanasia but at significantly lower incidence and severity indicating partial to complete recovery. (C) shows a minimal level of squamous metaplasia (arrow) in the respiratory epithelium of the nasal septum in level 2. (D) shows normal laryngeal respiratory epithelium from a control animal. (E) shows an area of expansion of the laryngeal squamous epithelium with attenuation and deciliation (arrow) and occasionally the presence of minimal inflammatory infiltrates (not present in this section). (F) Following the 28-day recovery period, etripamil-related microscopic observations were not present; the larynx is lined by normal laryngeal respiratory epithelium (hematoxylin-eosin, original magnification ×20).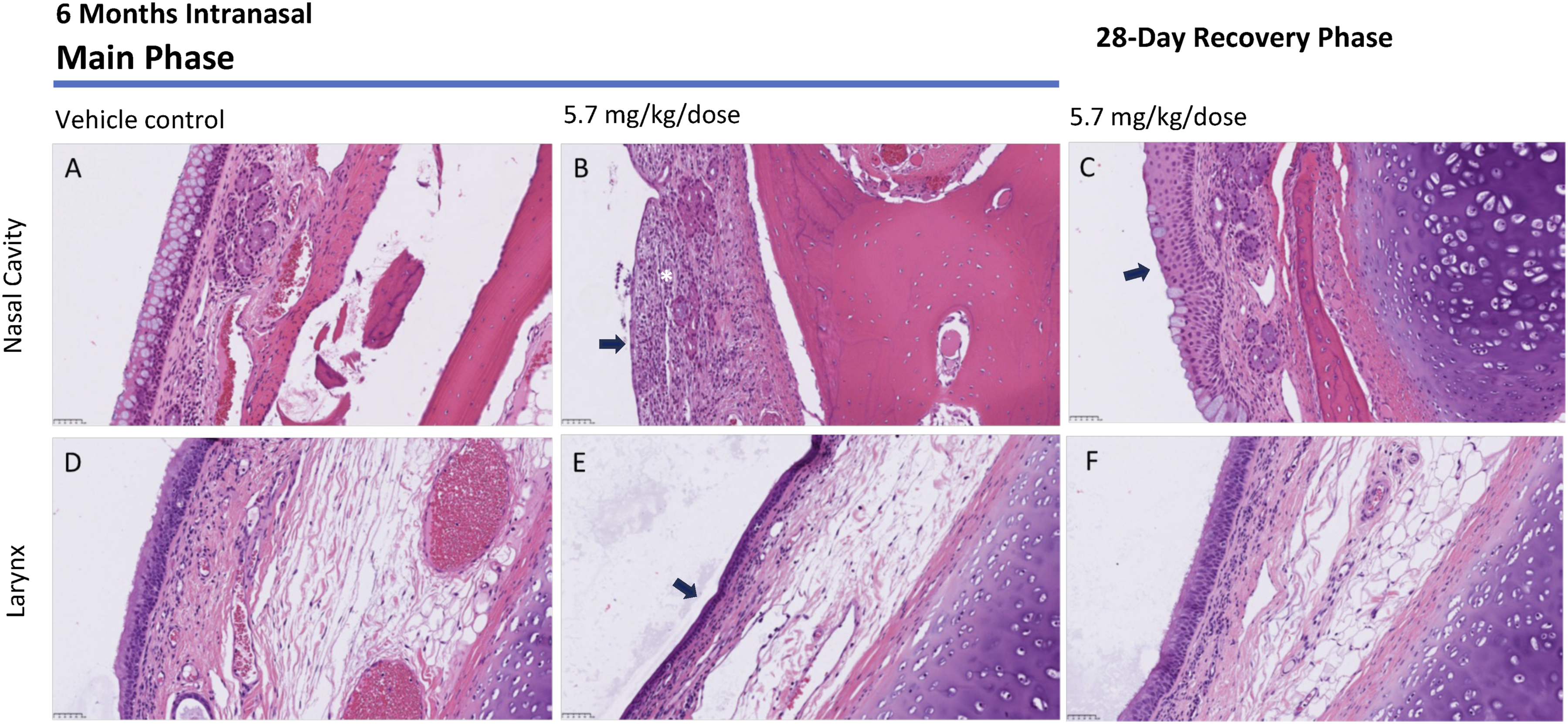
Summary of Mean Etripamil Toxicokinetic Parameters in Male and Female Cynomolgus Monkey Plasma Following Intranasal Administration on Days 1, 92, and 176.
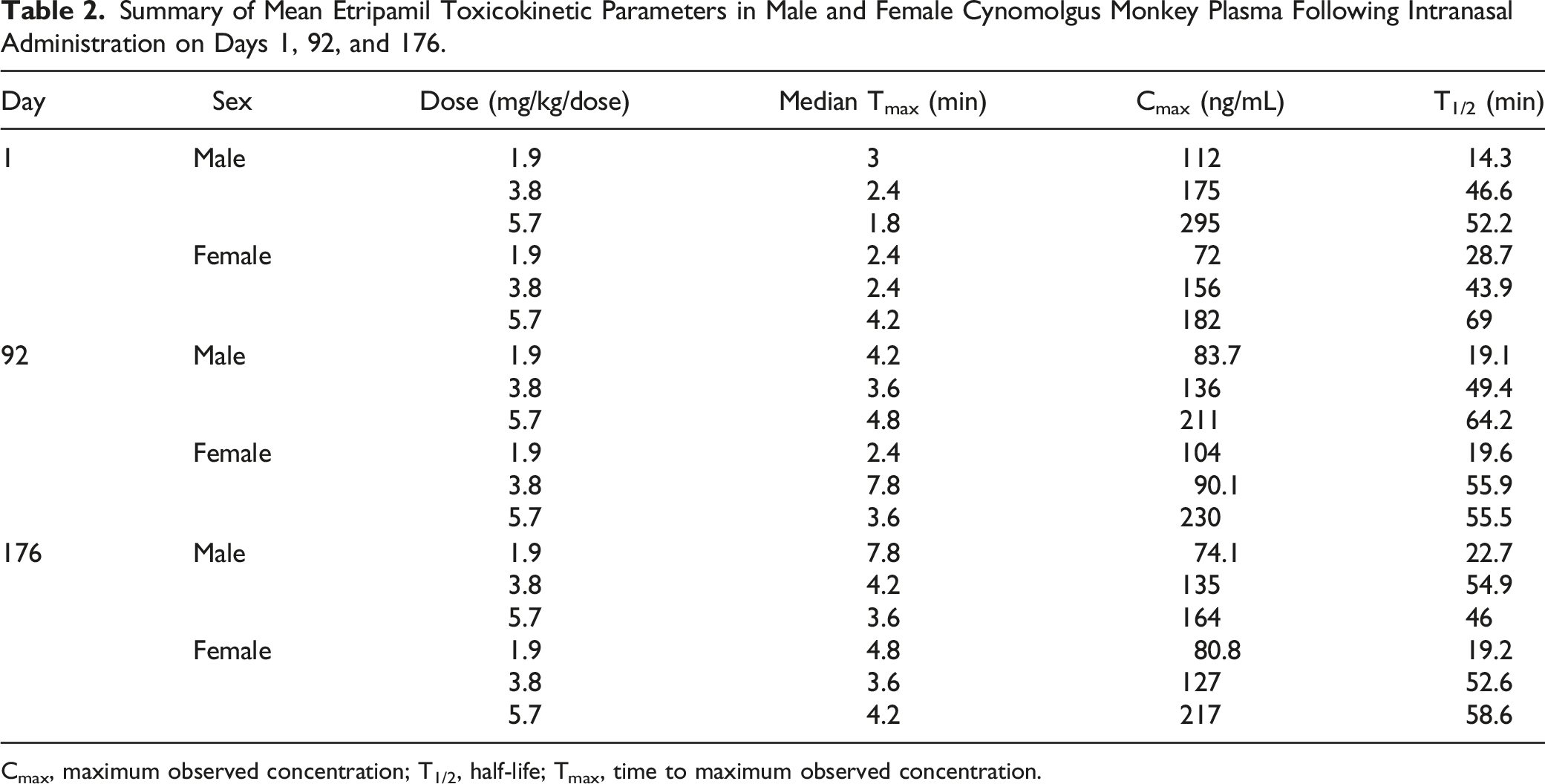
Cmax, maximum observed concentration; T1/2, half-life; Tmax, time to maximum observed concentration.
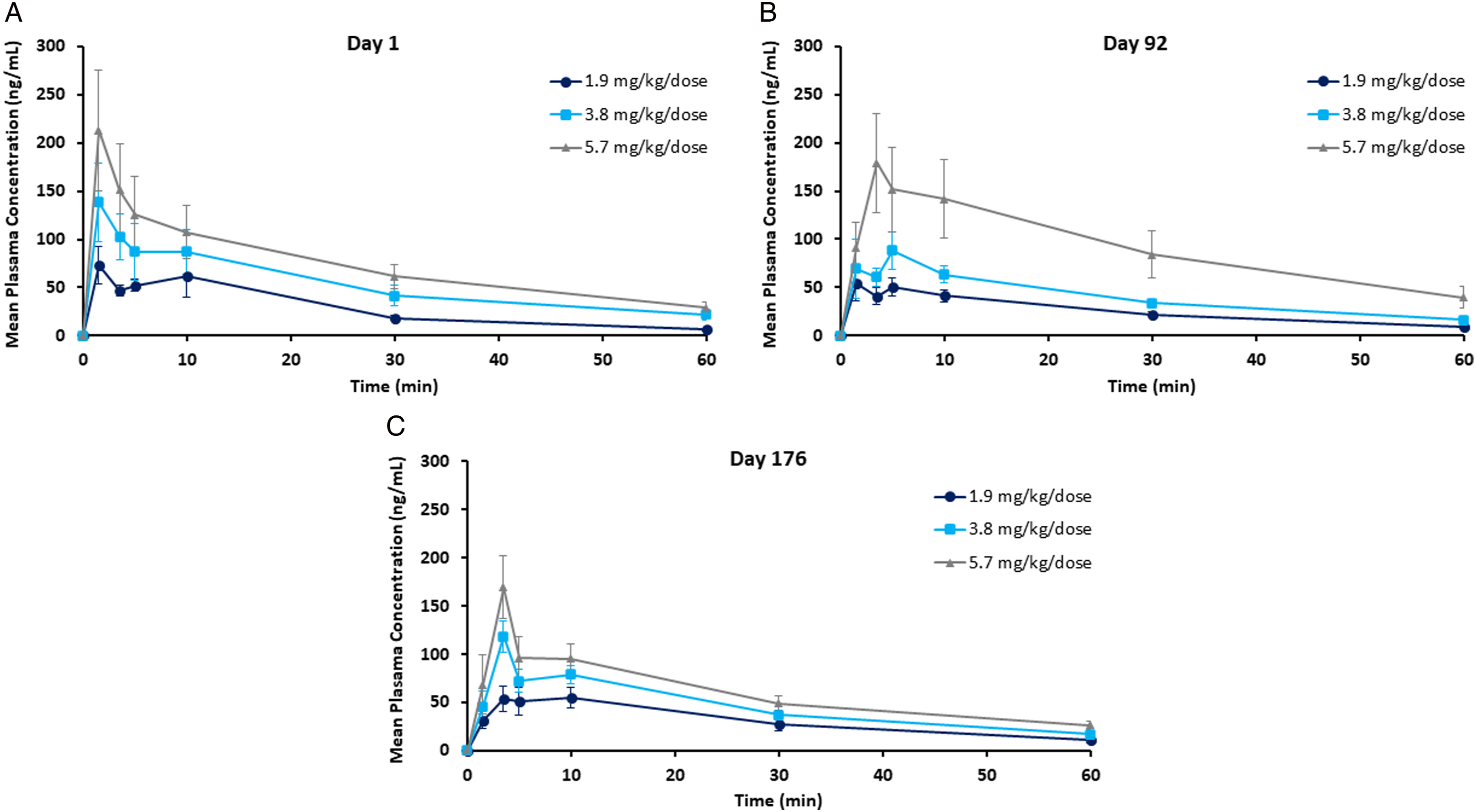
Summary of etripamil plasma concentrations following intranasal administration on Days 1, 92, and 176 in cynomolgus monkeys.
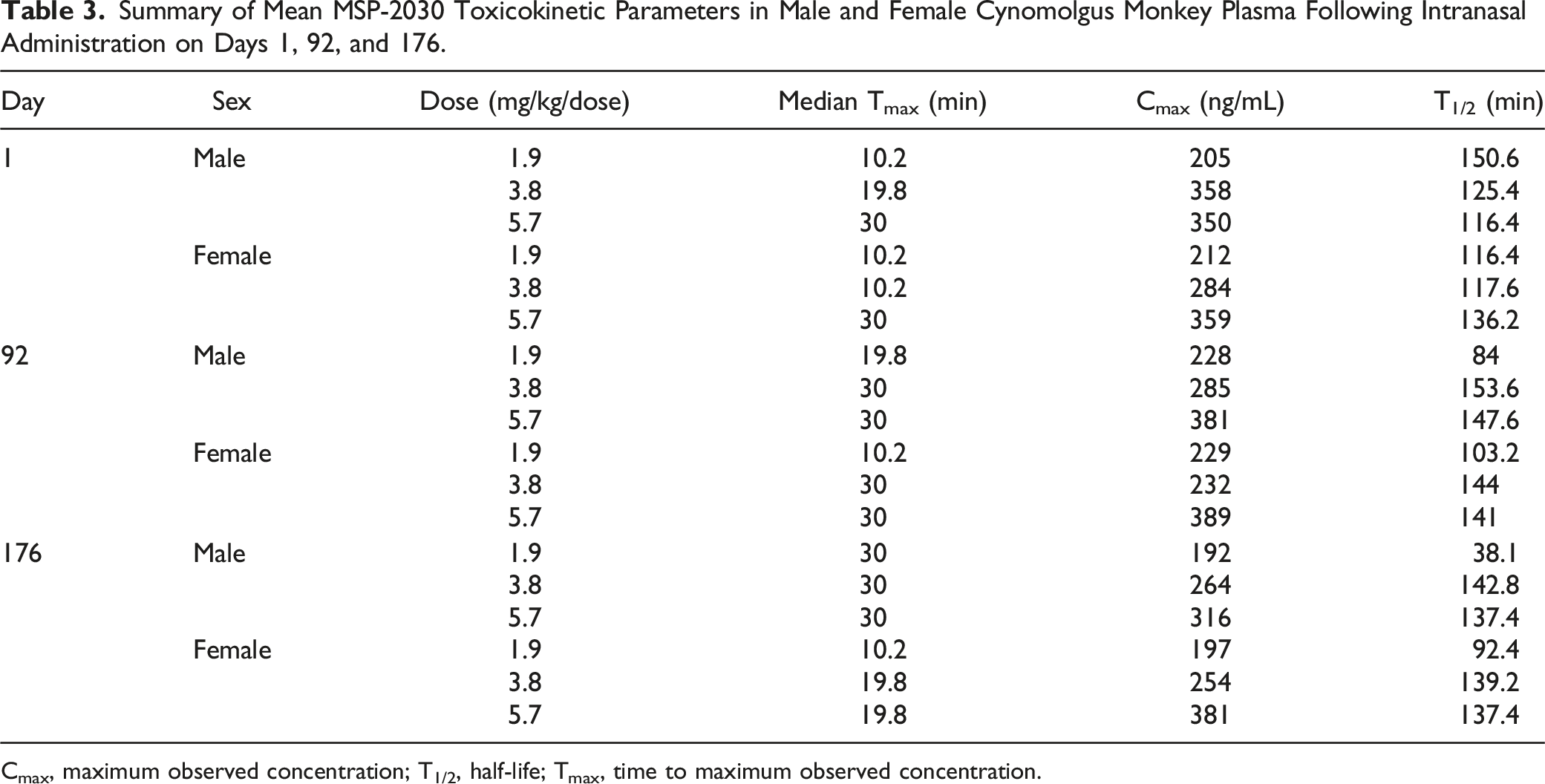
Following etripamil administration on Days 1, 92, and 176, plasma concentrations of MSP-2030 were observed up to 12 hours post dose for all the dose levels except for 2 animals at the 1.9 mg/kg/dose level, when plasma concentrations were observed up to 4 hours post dose on Days 92 and 76 (Figure 3). Following weekly administration of etripamil, maximum plasma concentrations of MSP-2030 were observed between 10 minutes and 1 hour post dose on Days 1, 92, and 176 for the dose values evaluated (Table 3). Maximum plasma concentrations of MSP-2030 were followed by a decline, with individual T1/2, when estimable, ranging from 38 minutes to 3.62 hours (Table 3). Summary of MSP-2030 plasma concentrations following intranasal administration on Days 1, 92, and 176 in cynomolgus monkeys. Summary of Mean MSP-2030 Toxicokinetic Parameters in Male and Female Cynomolgus Monkey Plasma Following Intranasal Administration on Days 1, 92, and 176. Cmax, maximum observed concentration; T1/2, half-life; Tmax, time to maximum observed concentration.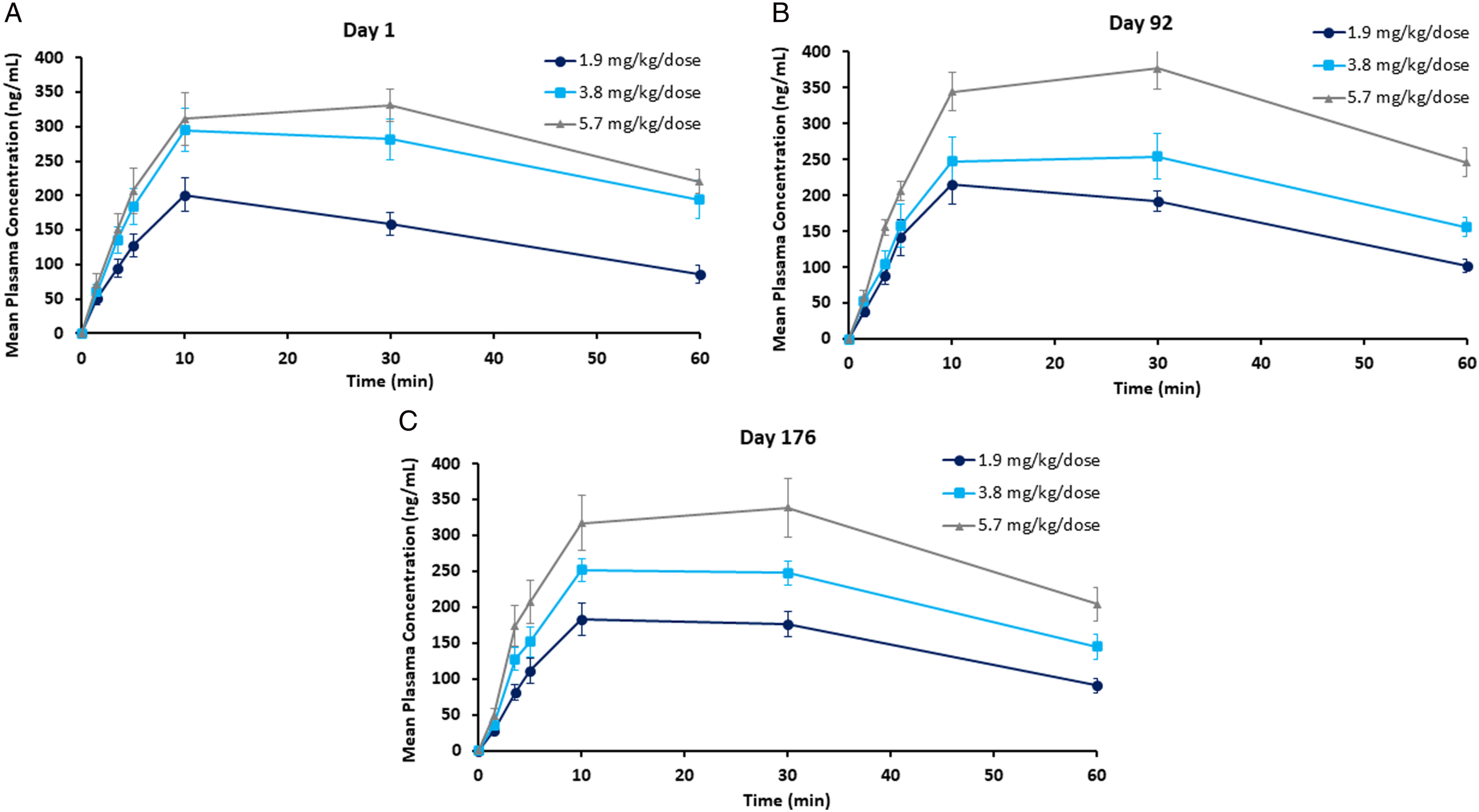
Discussion
Based on study findings, no mortality occurred at any dose. Histopathological changes in the nasal epithelium showed a dose-dependent trend with evidence of recovery after the 28-day recovery period. The systemic no observable adverse effect level (NOAEL) was determined to be the 5.7 mg/kg/dose, and the local toxicity NOAEL was the 1.9 mg/kg/dose. Systemic exposure to etripamil increased in a dose dependent manner following weekly administration. These data support the continued development of etripamil as an acute treatment of PSVT. Dose-dependent, histopathological changes were observed in the nasal cavity, larynx, and pharynx of cynomolgus macaques. These histopathologic changes were not indicative of systemic toxicity and there was evidence of recovery 28 days later.
It is well known that use of CCBs can result in systemic toxicity. Cardiac toxicity results from excessive negative inotropy and myocardial depression, negative chronotropy and sinus bradycardia, and negative dromotropy and AV node block. 10 Toxic effects of CCBs on vascular smooth muscle result in hypotension, shock, and coronary vasodilation. Metabolic effects of CCB toxicity can cause hypoinsulinaemia and CCB-induced insulin resistance. Pulmonary edema and renal failure may also occur. The oral route is the mainstay for administration of CCBs, which are metabolized by cytochrome P450 enzymes at first pass; at higher doses clearance of CCBs is decreased and changes from first order to zero order kinetics due to P450 saturation. 10 Results of the present study showing the minimal systemic toxicity of etripamil are therefore clinically important. Etripamil is locally intranasally administered and rapidly metabolized by plasma esterases due to the presence of an ester moiety in the molecule. The latter properties may contribute significantly to the lessened potential for systemic toxicity. Collectively, these data support the continued study of etripamil in humans as a potential treatment for PSVT.
Footnotes
Acknowledgments
Writing Assistance: Medical writing support was provided by Katie Crosslin, PhD, and Anushree Kogje, PhD, both of Two Labs Medical Affairs and Communications, which was in accordance with the Good Publication Practice guidelines.
Author Contributions
JP and CLM: methodology, validation, formal analysis, investigation, resources, writing-original draft, writing – review and editing, project administration. J-PM and VB: conceptualization, methodology, writing-original draft, writing – review and editing, visualization, project administration. DW: conceptualization, methodology, validation, writing-original draft, writing – review and editing, visualization, supervision, project administration.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Jean-Pierre Moreau is an employee of Recherche Continuum Research and was contracted by Milestone Pharmaceuticals to design and monitor the study. Charles River Laboratories was contracted by Milestone Pharmaceuticals to perform this study. Douglas Wight was an employee of Milestone Pharmaceuticals at the time of this study. Veronique Boulanger was an employee of Milestone Pharmaceuticals at the time of this study.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by Milestone Pharmaceuticals (Charlotte, NC, USA; Montréal, Québec, Canada).
Ethical Statement
Data Availability Statement
Qualified researchers from an appropriate institution may request access to data that underlie the results reported in this article (text, tables, figures, and appendices). Upon approval of a data sharing request, information necessary to address the research question will be provided under the terms of a signed data sharing agreement. Requests should be submitted to